一种制备3-烯基吡咯衍生物的方法与流程
本发明涉及一种制备3-烯基吡咯衍生物的方法。具体方法是由简单制备得到的氮杂烯炔化合物在碱存在的条件下环化制备3-烯基吡咯。
背景技术:
吡咯及其衍生物是重要的杂环化合物之一,其中包含与芳香环共轭的烯烃的烯基吡咯类化合物,广泛应用于血色素和叶绿素等具有生理活性的化合物或天然产物的全合成中。除此之外,还可通过D-A反应合成吲哚类化合物,同时还可通过自聚合成非线性的光材料。近年来,烯基吡咯在光感应器及新的光催化剂的配体方面得到了很大关注。(文献1:(a)Jones,R.A.;Bean,G.P.The Chemistry of Pyrroles;Academic Press:New York,1989;(b)Hodges,L.M.;Moody,M.W.;Harman,W.D.J.Am.Chem.Soc.1994,116,7931;(c)Sour,A.;Rurack,K.Kollmannsberger,M.;Daub,J.Angew.Chem.,Int.Ed.2001,40,385;(d)Boillot,M.L.;Riviere,E.;Lesot,P.Eur.J.Inorg.Chem.1999,12,2117.)
因此,烯基吡咯类化合物的合成方法很值的进一步的研究。然而对于烯基吡咯的合成方法存在很多缺点,比如通过吡咯合成的方法(式1,a),合成步骤长;通过不饱和肟合成的方法(式1,b),反应选择性差;Pd催化的反应(式1,c),产物仅为单取代烯烃等。(文献2:(a)Salvadori,P.;Settambol,R.;Lazzaron,R.;Messer,T.;Mazzett,M.J.
式1
J.Org.Chem.1993,58,7899;(b)Mikhaleva,A.I.;Sigalov,M.V.;Halabin,G.A.Tetrahedron Lett.1982,23,5063;(c)Scott,A.I.;Wang,J.J.Tetrahedron Lett.1955,36,7043.),本专利则是以氮杂烯炔为原料,在碱的作用下环化得到3-烯基吡咯衍生物。该方法原料廉价易得,操作简便
技术实现要素:
为解决上述问题,本发明提供了一种制备3-磺酰基取代的吡咯衍生物的方法,技术方案如下:
以N-磺酰基取代的1,5-烯炔1为原料,合成3-烯基吡咯衍生物2,反应式如下:
其中R、R1、R2、R3中的任一或二种以上为C1-C8烷基、C1-C8烷氧基、萘基、呋喃基、苯基、或取代的苯基中的任意一种或二种以上,上述取代的苯基中,苯基上的取代基为C1-C8烷基、C1-C8烷氧 基、CF3、F、Cl、Br、I、NO2中的一种或二种以上。
具体操作步骤如下:
在惰性气体环境中,N-磺酰基取代的1,5烯炔1中加入无机碱,40℃-120℃下反应24h-48h,反应结束后,用旋转蒸发仪蒸干溶剂,所得固体即为3-烯基吡咯类化合物2。
无机碱为碳酸铯,叔戊酸铯,氢氧化钾,碳酸钾等其中的一种或两种。
无机碱的用量为底物的2当量。
所述溶剂为乙腈、二氯甲烷、二氯乙烷、N,N-二甲基甲酰胺、甲苯、乙醚、四氯化碳、四氢呋喃或1,4-二氧六环中的一种或二种以上;溶剂与底物的用量比例为10ml/mmol-30ml/mmol。
反应温度范围为40℃-120℃,反应时间为8h-16h。
所述惰性气体为氩气;反应结束后,用旋转蒸发仪蒸干溶剂,温度25℃<<40℃,所得固体可溶于二氯甲烷上样进行硅胶柱层析进行产物分析。
本发明有以下优点:
1.反应物氮杂1,5-烯炔1由廉价易得的原料醛、磺酰胺和炔经简单反应步骤得到。
2.生成的3-烯基吡咯衍生物2的反应操作简单,只通过简单的一步反应就可构建吡咯杂环,不需要进行多步反应。
附图说明
图1为实施例1底物1a的核磁氢谱;
图2为实施例1底物1a的核磁碳谱;
图3为实施例1底物1a的高分辨谱图;
图4为实施例1底物2a的核磁氢谱;
图5为实施例1底物2a的核磁碳谱;
图6为实施例1底物2a的高分辨谱图;
图7为实施例1制得的2a的晶体衍射图。
具体实施方式
(1)参照文献,经三步合成氮杂1,5-烯炔:第一步,芳香醛和磺酰胺在原硅酸乙酯中反应生成亚胺(文献3:Love,B.E.;Raje,P.S.;Williams II,T.C.Synlett 1994,493.)(式2,反应方程式1);当醛为脂肪醛时,将醛、磺酰胺和对甲苯亚磺酸钠溶在甲酸和水中反应,得到脂肪族亚胺(文献4:Chemla,F.;Hebbe,V.;Normant,J.F.Synthesis2000,75.)( 式2,反应方程式2)。第二步,在低温下将端炔进行锂化,然后将亚胺的四氢呋喃溶液滴加到反应体系中得到产物炔丙胺3(文献5:Katritzky,A.R.;Li,J.Q.;Gordeev,M.F.Synthesis 1994,93.)( 式2,反应方程式3)。第三步,以二氯甲烷为溶剂,碳酸铯作为 催化剂,炔丙胺和DMAD发生加成反应生成氮杂1,5-烯炔1。(式2,反应方程式4)。(文献6:Wan,B.S.;Xin,X.Y.Angew.Chem.Int.Ed.2012,51,1693.)
式2.氮杂1,5-烯炔的合成步骤
(2)由氮杂1,5-烯炔制备3-烯基吡咯衍生物
式3.3-烯基吡咯衍生物的合成步骤
反应方程式见式3,于反应器中进行反应,反应器与手套箱中,完全氩气环境中,加入0.2mmol的氮杂1,5-烯炔1,1.2eq.的碳酸铯,然后加入2mL溶剂,40℃-120℃下反应8h-16h。反应结束后旋蒸掉溶剂后,固体进行硅胶柱层析,得到3-烯基吡咯衍生物2。
实施例1
于10ml封管中进行反应,加入0.3mmol(145.1mg)的1a,3.6mmol(117mg)的碳酸铯,然后加入3mlTHF,80℃下搅拌反应12小时。反应结束后,用旋转蒸发仪抽掉溶剂后,固体溶于2mL二氯甲烷上样进行硅胶柱(300-400目硅胶)层析,用石油醚:乙酸乙酯=5:1的洗脱剂冲洗柱子,旋蒸浓缩得到固体,最终得到78.5mg的吡咯衍生物2a,分离收率为80%。
核磁数据均来自于Bruker 400MHz核磁,核磁氢谱内标为TMS,CDCl3为7.26ppm,碳谱CDCl3为77.16ppm。高分辨仪器为四级杆-飞行时间质谱仪。
1a的表征数据如下:
1H NMR(400MHz,CDCl3)δ7.90(d,J=8.3Hz,2H),7.56(d,J=7.9Hz,2H),7.37–7.26(m,5H),6.29(s,1H),6.04(s,1H),3.76(s,3H),3.58(s,3H),2.45(s,3H),2.09–1.97(m,2H),1.35–1.24(m,4H),0.87(t,J=7.0Hz,3H).(谱图见说明书附图1)。
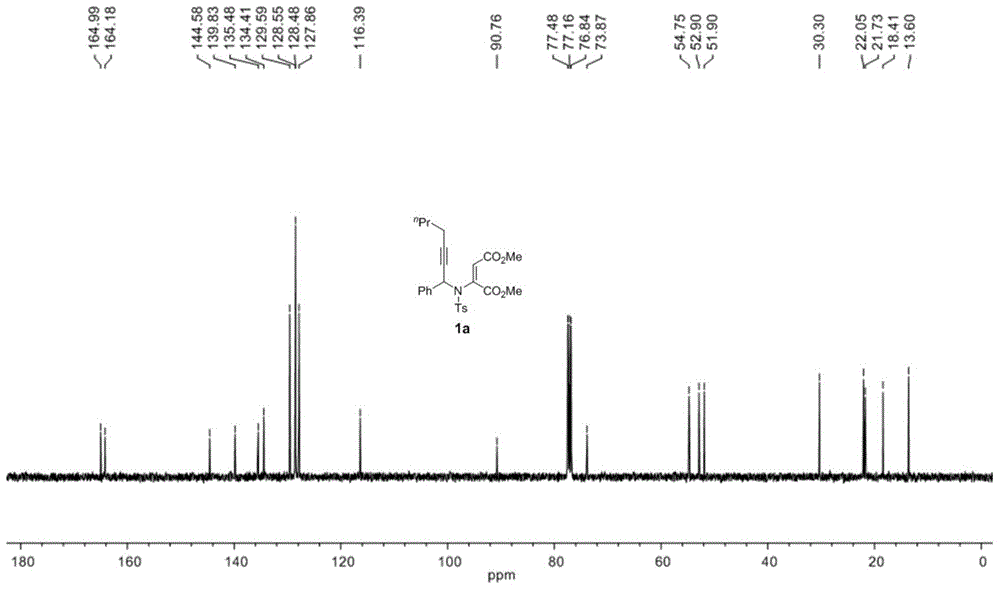
13C NMR(100MHz,CDCl3)δ165.0,164.2,144.6,139.8,135.5,134.4,129.6,128.6,128.5(2),127.9,116.4,90.8,73.9,54.8,52.9,51.9,30.3,22.1,21.7,18.4,13.6.(谱图见说明书附图2)。
HRMS calcd for C26H30NO6S[M+H]+484.1794,found 484.1778.(见说明书附图3)。
2a的表征数如下:
1H NMR(400MHz,CDCl3)δ9.54(s,1H),7.48(d,J=7.1Hz,2H),7.42(t,J=7.2Hz,2H),7.39–7.33(m,1H),6.28(d,J=16.0Hz,1H),5.90–5.78(m,1H),3.91(s,3H),3.78(s,3H),2.13–2.04(m,2H),1.46–1.35(m,2H),0.90(t,J=7.4Hz,3H).(谱图见说明书附图4)
13C NMR(100MHz,CDCl3)δ167.2,160.8,133.4,131.3,128.9,128.8,128.4,128.3,121.3,120.4,120.0,119.9,52.5,52.2,35.6,22.6,13.7.(谱图见说明书附图5)。
HRMS calcd for C19H22NO4[M+H]+328.1549,found 328.1552.(见说明书附图6)。
- 还没有人留言评论。精彩留言会获得点赞!