一种水溶性阳离子亚苄基环烷烃酮光敏剂及其制备方法与在光动力灭菌中的应用与流程
本发明涉及光动力治疗领域。更具体地,涉及一种水溶性阳离子亚苄基环烷烃酮光敏剂的制备方法及其在光动力灭菌中的应用。
背景技术:
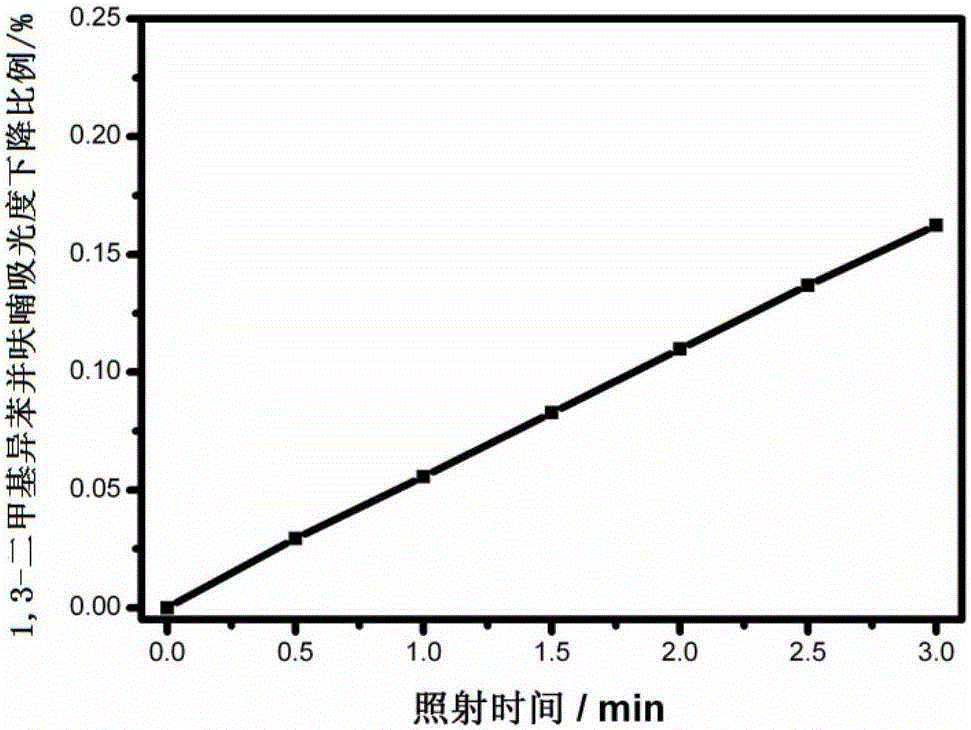
:抗生素的发现曾使人类一度认为即将告别微生物感染的时代。但是,随着抗生素的普遍应用和不断升级,临床致病菌感染并未得到完全控制,与之相反,许多致病菌或条件致病菌在生存环境的压力下,变异成了拥有有效抵御抗生素杀伤耐药机制的多重耐药菌。多重耐药菌引起的感染导致的高死亡率和高医疗费使抗微生物感染的工作面临着严峻的考验。2010年,英国医学期刊《柳叶刀》公布的携带“新德里金属蛋白酶-1(NDM-1)”基因的“超级细菌”几乎可以耐受所有的抗生素,但是目前临床上常用的抗感染药物和方法对此类感染的疗效十分有限。因此,一旦出现具有区域感染能力的泛耐药菌感染,后果将十分严重。光动力治疗(photodynamictherapy,PDT)是一种利用光敏剂在光的作用下可以产生消灭病变细胞或细菌的活性物质,从而达到治疗目的的治疗手段。光动力疗法抗微生物感染已在研究之中,体外研究已证明此方法可有效对抗微生物(包括耐药菌),且天然和人工合成的光敏剂均可使用。近期,Demidova和Hamblin等人在“PhotodynamicinactivationofBacillussporesmediatedbyphenothiaziniumdyes”,ApplEnvironMicrobiol,2005,vol.71,pp6918-6925的文章中也证实了,在吩噻嗪类染料存在的温和条件下,利用红光照射细菌芽孢(对大多数抗菌剂有耐药性)培养体系可使芽孢杆菌灭活。JoriG.等人在“Photodynamictherapyinthetreatmentofmicrobialinfections:basicprinciplesandperspectiveapplications”,LasersinSurgeryandMedicine,2006,vol.38,pp468-481的文章中指出在生理pH值下,阳离子型光敏剂如吩噻嗪衍生物、酞菁衍生物、卟啉衍生物能够有效导致革兰氏阴性和革兰氏阳性细菌的灭活,另外其对酵母菌、真菌、支原体和致病原虫也有良好的光动力疗效。与传统的抗生素治疗相比,光动力灭菌具有以下主要优势:(1)、广 泛的抗菌谱,对细菌、真菌、病毒、原虫等均有效,对多重耐药菌株同样有效;(2)、能够选择性的杀伤病原微生物,而不损伤宿主组织;(3)、不会导致菌株的耐药性;(4)、光敏剂的毒副作用小,对肝肾功能的损伤小。实现病原微生物的有效灭活同时将PDT对宿主细胞和组织的损伤降到最低是光动力灭活病原微生物的理想结果。Friedberg等人在“Antibody-TargetedPhotolysis:BacteriocidalEffectsofSn(IV)Chlorine6-Dextran-MonoclonalAntibodyConjugates”,AnnalsoftheNewYorkAcademyofSciences,1991,vol.66,pp383-393的文章中提出了在抗菌剂上连接靶向菌体的化学分子的方法,取得了较好的效果。但是,此类方法会使抗菌剂的广谱性显著降低。因此,实现PDT选择性的更优策略仍需探索。前期研究表明,哺乳动物细胞膜和微生物的细胞膜的成分和结构存在显著差异,其中,微生物细胞膜外层呈负电性,而哺乳动物细胞膜外层近乎电中性。基于以上特点,设计、合成在生理pH值下呈现正电性的光敏剂(阳离子光敏剂)对于实现光动力灭活的选择性至关重要。另外,阳离子光敏剂除了能够利用活性氧物质灭活病原微生物外,还可以通过扰乱和中断微生物有序的细胞膜结构发挥抗菌效果。由于阳离子光敏剂特殊的抗菌机制,因此,它被认为具有抗菌广谱性。目前已有的临床光动力治疗用光敏剂如血卟啉衍生物HpD、photofrin、verteporfin等,这些光敏剂均存在不同程度的有效组份不清,生物体内的吸收代谢机理不明确,代谢较差等问题。而用于临床实践阶段的光敏剂吩噻嗪类染料亚甲蓝(methyleneblue,MB),在细菌表面极易形成二聚体,严重影响了光动力灭活的效率。基于以上问题,需要设计、合成一些化学结构明确、溶解性好且对病原微生物具有有效杀伤作用的新型光敏剂。技术实现要素:本发明的一个目的在于提供一种水溶性阳离子亚苄基环烷烃酮光敏剂。该类光敏剂具有确定的化学结构且分子结构简单,在水中溶解度良好,具有合适的脂水分配比,且在350~600nm波长范围内有较强吸收,在光源照射下能够快速产生活性氧物质,可用于光动力灭菌。本发明的第二个目的在于提供一种水溶性阳离子亚苄基环烷烃酮光敏剂的制备方法。提供了一种可光动力选择性灭活病原微生物而不损伤宿主细胞的水溶性阳离子亚苄基环烷烃酮光敏剂的设计思路。该方法操作简单、反应 条件温和、纯化过程简便、产品纯度可达到药物要求的特点。本发明的第三个目的在于提供一种水溶性阳离子亚苄基环烷烃酮光敏剂的用途。为达到上述第一个目的,本发明采用下述技术方案:一种水溶性阳离子亚苄基环烷烃酮光敏剂,其特征在于,包括具有如下通式C1、C2或C3结构的化合物:其中:R2,R3和R4相同或不同,R2、R3和R4选自甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基或-(CH2)n-R1X;优选地,R2,R3和R4选自甲基、乙基或-(CH2)n-R1X;R5和R6相同或不同,R5和R6选自N或P;优选地,R5和R6为N;X为负离子Cl-、Br-或I-;n=1、2、3或4;优选地,n为1或2;n1和n2=0、1、2、3或4;优选地,n1和n2为0、1或2;R1为季铵盐阳离子;优选地,R1选自A选自N或P;优选地,A为N;A1,A2和A3相同或不同,A1、A2和A3选自甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基或苯基;优选地,A1,A2和A3选自甲基、乙基或苯基;选自环丁酮、环戊酮、环己酮、环庚酮或环辛酮。为达到上述第二个目的,本发明一种水溶性阳离子亚苄基环烷烃酮光敏剂的制备方法,具有通式C1结构的化合物包括如下合成步骤:1)将五氯化磷与二甲基甲酰胺反应后,再与通式Q1化合物反应,收集得到通式Q2中间产物2)在乙醇-二氯甲烷溶液的碱性催化剂条件下,将与所得通式Q2中间产物反应,收集得到通式Q3中间产物,3)在乙醇-二氯甲烷溶液的碱性催化剂条件下,将所得通式Q3中间产物与通式Q4化合物反应;或者,在乙醇-二氯甲烷溶液的碱性催化剂条件下,将与所得通式Q2化合物反应;收集得到的通式Q5中间产物4)在溶剂为二甲基甲酰胺(DMF)条件下,将所得通式Q5中间产物与叔胺或氮杂环化合物反应,收集得到通式C1结构的化合物;其中:n3和n4相同或不同,n3、n4=1、2、3或4;优选地,n3、n4为1或2;n5和n6相同或不同,n5、n6=1、2、3或4;优选地,n5和n6为1或2;B1和B2相同或不同,B1和B2选自-H、-OH、-Cl、-Br或-I;B3选自N或P;优选地,B3为N;B4、B5选自-H、-Cl、-Br或-I;B4、B5相同或不同,但至少有一个选自-Cl、-Br或-I;B6选自N或P;优选地,B6为N;B7和B8选自-H、-Cl、-Br或-I;B7和B8相同或不同,但至少有一个选自-Cl、-Br或-I;选自环丁酮、环戊酮、环己酮、环庚酮或环辛酮。优选地,具有通式C1结构的化合物的合成步骤2)、3)中所述碱性催化剂为氢氧化锂、氢氧化钠、氢氧化钾、无水碳酸钠、无水碳酸钾、吡啶或六氢吡啶中的一种或几种的组合。为达到上述第二个目的,本发明一种水溶性阳离子亚苄基环烷烃酮光敏 剂的制备方法,具有通式C2结构的化合物包括如下合成步骤:1)将五氯化磷与二甲基甲酰胺反应后,再与通式S1化合物反应,收集得到通式S2中间产物2)在乙醇-二氯甲烷溶液的碱性催化剂条件下,将与所得通式S2中间产物反应,收集得到通式S3中间产物3)在乙醇-二氯甲烷溶液的碱性催化剂条件下,将所得通式S3中间产物与通式Q2化合物反应;或者,在乙醇-二氯甲烷溶液的碱性催化剂条件下,将所得通式S2化合物与通式Q3化合物反应,收集得到的通式S4中间产物4)在溶剂为二甲基甲酰胺(DMF)条件下,将所得通式S4中间产物与叔胺或氮杂环化合物反应,收集得到通式C2结构的化合物;其中:n1=0、1、2、3或4;优选地,n1为0、1或2;n3和n4相同或不同,n3、n4=1、2、3或4;优选地,n3、n4为1或2;D1选自-H、-OH、-Cl、-Br或-I;D2选自-H、-Cl、-Br或-I;B3选自N或P;优选地,B3为N;B4、B5选自-H、-Cl、-Br或-I;B4、B5相同或不同,但至少有一个选自-Cl、-Br或-I;选自环丁酮、环戊酮、环己酮、环庚酮或环辛酮。优选地,具有通式C2结构的化合物的合成步骤2)、3)中所述碱性催化剂为氢氧化锂、氢氧化钠、氢氧化钾、无水碳酸钠、无水碳酸钾、吡啶或六氢吡啶中的一种或几种的组合。为达到上述第二个目的,本发明一种水溶性阳离子亚苄基环烷烃酮光敏剂的制备方法,具有通式C3结构的化合物包括如下合成步骤:1)将通式S3化合物与通式S5化合物在乙醇-二氯甲烷溶液的碱性催化剂条件下反应;或者,将通式S2与通式在乙醇-二氯甲烷溶液的碱性催化剂条件下反应;收集得到通式T1中间产物2)在溶剂为二甲基甲酰胺(DMF)条件下,将所得通式T1中间产物与叔胺或氮杂环化合物反应,收集得到通式C3结构的化合物;其中:n1=0、1、2、3或4;优选地,n1为0、1或2;n2=0、1、2、3或4;优选地,n2为0、1或2;D2选自-H、-Cl、-Br或-I;D3选自-H、-Cl、-Br或-I;D3和D2相同或不同,但至少有一个选自-Cl、-Br或-I;选自环丁酮、环戊酮、环己酮、环庚酮或环辛酮。优选地,具有通式C3结构的化合物的合成步骤1)中所述碱性催化剂为氢氧化锂、氢氧化钠、氢氧化钾、无水碳酸钠、无水碳酸钾、吡啶或六氢吡啶中的一种或几种的组合。为达到上述第三个目的,本发明的水溶性阳离子亚苄基环烷烃酮光敏剂应用在光动力灭活细菌、真菌和病毒。优选地,所述细菌包括按照革兰氏染色分类的革兰氏阳性菌和革兰氏阴性菌;更优选地,所述革兰氏阳性菌包括金黄色葡萄球菌、链球菌、肺炎双球菌、炭疽杆菌、白喉杆菌或破伤风杆菌;所述革兰氏阴性菌包括大肠杆菌、痢疾杆菌、伤寒杆菌、变形杆菌或百日咳杆菌。优选地,所述真菌包括霉菌、酵母菌、白色念珠菌、啤酒母菌、红曲霉素、假丝酵母、黄曲霉、白地霉或抗生菌。优选地,所述病毒包括按照寄主类型分类的细菌病毒、植物病毒、动物病毒;优选地,所述细菌病毒包括噬菌体;优选地,所述植物病毒包括烟草花叶病毒;优选地,所述动物病毒包括HIV、天花病毒、甲型肝炎病毒、乙型肝炎病毒或风疹病毒。本发明的水溶性阳离子亚苄基环烷烃酮光敏剂应用在光动力灭活细菌、真菌和病毒的机制为阳离子抗菌机制,阳离子光敏剂和菌体之间特殊的静电相互作用,从而中断和扰乱细菌膜结构是其抗菌机制中的重要部分。本发明的有益效果如下:1.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂结构简单、分子量小,具有确定的化学结构,易于制备、纯化和进一步修饰,满足临床用药的基本要求。2.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂具有脂水双亲的特点,其脂水分配比能够满足临床光动力治疗的使用要求。3.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂的合成方法具有操作简单、产品产率高、纯度高的特点,可以放量合成。4.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂在350~600nm波长范围具有较高的生物光动力活性,作为光动力药物灭活病原微生物方面具有很好应用前景。5.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂能够实现细菌、真菌和病毒的有效光灭活,具有广谱抗菌性,补充了亚苄基环烷烃酮抗菌领域很大的空白。6.本发明中的水溶性阳离子亚苄基环烷烃酮光敏剂能够选择性的灭活病原微生物而尽可能的减少对宿主细胞的损伤,这一效果强有力的证明了此类阳离子光敏剂应用于临床的光明前景。附图说明下面结合附图对本发明的具体实施方式作进一步详细的说明。图1示出实施例1中光敏剂C1-1在二甲基甲酰胺(DMF)和磷酸缓冲盐溶液(PBS)缓冲液中的吸收光谱。图2示出实施例1中光敏剂C1-1存在下,1,3-二甲基异苯并呋喃(DPBF)在DMF中的吸光度随时间的下降比例。图3示出实施例7中光敏剂C1-2在DMF和PBS缓冲液中的吸收光谱。图4示出实施例7中光敏剂C1-2存在下,DPBF在DMF中的吸光度随时间的下降比例。图5示出实施例12中光敏剂C1-3在DMF和PBS缓冲液中的吸收光谱。图6示出实施例12中光敏剂C1-3存在下,DPBF在DMF中的吸光度随时间的下降比例。图7示出光动力灭活金黄色葡萄球菌、大肠杆菌的菌板光敏剂浓度示意图。图8示出实施例40的实验结果:实施例2中制得的光敏剂用于光动力灭活金黄色葡萄球菌的实验。图9示出实施例41的实验结果:实施例2中制得的光敏剂用于光动力灭活大肠杆菌的实验。图10示出实施例42的实验结果:实施例2中制得的光敏剂用于光动力灭活白色念珠菌的实验。图11示出实施例43的实验结果:实施例8中制得的光敏剂用于光动力灭活金黄色葡萄球菌的实验。图12示出实施例44的实验结果:实施例8中制得的光敏剂用于光动力灭活大肠杆菌的实验。图13示出实施例45的实验结果:实施例8中制得的光敏剂用于光动力灭活白念珠菌的实验。图14示出实施例46的实验结果:实施例13中制得的光敏剂用于观察细胞摄取阳离子光敏剂的实验。具体实施方式为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。附图中相似的部件以相同的附图标记进行表示。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。实施例1一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间体化合物Q2-1的制备:向冰浴冷却的150mL的DMF中缓慢加入52.06克(0.25mol)五氯化磷,继续冰浴30分钟后,再室温搅拌15分钟。向上述体系中缓慢加入N-乙基-N-羟乙基苯胺20克(0.12mol),控温45℃搅拌12小时后,恢复至常温并用碳酸氢钠溶液中和反应液。反应液用二氯甲烷萃取三次,二氯甲烷萃取液用水洗至中性后加入无水硫酸镁干燥,经过滤、旋蒸除去二氯甲烷,得到24.9克相应的中间产物Q2-1(产率98%)。(II)中间体化合物Q3-1的制备:向一个100毫升三口烧瓶中加入10.58克(0.05mol)Q2-1,3.52克(0.05mol)环丁酮,40毫升二氯甲烷和20毫升乙醇,搅拌使其溶解均匀后一次性加入0.03克氢氧化钠,20℃下反应10小时,反应液经旋蒸除去溶剂得到粗产品,用色谱柱分离提纯得5.9克(产率44%)橙色固体Q3-1。(III)中间体化合物Q5-1的制备:向一个100毫升三口烧瓶中加入2.4克(0.01mol)Q3-1,1.8克(0.01mol)商业可得的4-(N,N-二乙氨基)苯甲醛,40毫升二氯甲烷和10毫升乙醇,搅拌使其溶解均匀后一次性加入0.06克氢氧化钠,室温下搅拌12小时,经旋蒸除去溶剂得到粗产品,用色谱柱分离提纯得到Q5-1固体1.5克(产率35%)。(IV)目标产物C1-1的制备:向一个100毫升三口烧瓶中加入0.23克(0.5mmol)Q5-1,1.96克吡啶,10毫升DMF,搅拌使其溶解均匀后加热100℃反应12小时。待反应体系冷却到室温后,利用甲苯重结晶得到C1-1固体80毫克(产率30%)。HR-MS(ESI):[M]+:Calcdfor[C31H36N3O]+466.2852;found466.2861。(V)用pH值7.4的磷酸盐缓冲液(简称:PBS缓冲液)检测目标光敏剂C1-1在水体系中的溶解度,25℃其溶解度大于4mg/mL;将目标光敏剂C1-1分别溶于DMF和PBS缓冲液中,测试其吸收光谱,证明了光敏剂C1-1在350~600nm波长范围有较强吸收峰,见图1。(VI)将上述目标光敏剂C1-1溶于DMF中,使其在473nm的吸光度为0.1,以酞菁锌(单线态氧量子产率ΦΔ=0.56)为参比,利用DPBF为活性氧 的捕获剂,在空气饱和的条件下完成活性氧量子产率的测定,结果如图2所示。说明光敏剂C1-1在473nm激光激发条件下产生了活性氧物质。实施例2重复实施例1,其不同在于,为环辛酮,催化剂为无水碳酸钠,以(2,2-联吡啶)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率34%。实施例3重复实施例1,其不同在于,为环庚酮,催化剂为无水碳酸钾,以(2-苄基吡啶)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率45%。实施例4重复实施例1,其不同在于,为环己酮,催化剂为氢氧化钾,以(二吡啶基乙烷)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率24%。实施例5重复实施例1,其不同在于,以(7,8-苯并喹啉)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率55%。实施例6重复实施例1,其不同在于,以(4,7-二氮杂菲)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率33%。实施例7一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间体化合物Q2-2的制备:参照实施例1中(I)的操作,用摩尔量1:3的N,N-二羟乙基苯胺和vilsmeier试剂反应,得到中间产物Q2-2(产率97%)。(II)中间体化合物Q3-2的制备:参照实施例1中(II)的操作,用摩尔量为1:1的Q2-2与环丁酮反应,催化剂为氢氧化钾,得到中间产物Q3-2(产率:39%)。(III)中间体化合物Q5-2的制备:参照实施例1中(III)的操作,用摩尔量1:1的Q3-2和4-(N,N-二乙氨基)苯甲醛反应,催化剂为氢氧化钾,得到中间产物Q5-2(产率62%)。(IV)目标产物C1-2的制备:参照实施例1中(IV)的操作,用质量比为1:10的Q5-2与吡啶反应,得到目标光敏剂C1-2(产率:90%)。HR-MS(ESI):[M]+:Calcdfor[C36H40ClN4O]+579.2885;found579.2889。(IV)参照实施例1中(V)的操作,证明C1-2溶解度大于5mg/mL;且在350~600nm波长范围有较强吸收峰,见图3。(V)参照实施例1(VI)的操作,结果同样证明了目标光敏剂C1-2在473nm激光照射下能产生活性氧物质,见图4。实施例8重复实施例7,其不同在于,为环己酮,催化剂为无水碳酸钠,以(喹啉)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率88%。实施例9重复实施例7,其不同在于,为环庚酮,催化剂为无水碳酸钠,以(二苯基-2-吡啶甲烷)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率77%。实施例10重复实施例7,其不同在于,以(2,2-联喹啉)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率47%。实施例11重复实施例7,其不同在于,以三乙胺代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率87%。实施例12一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间产物Q5-3的制备:参照实施例1中(III)的操作,用摩尔量为1:2的环丁酮与Q2-1反应,催化剂为吡啶,得到中间产物Q5-3(产率:85%)。(II)目标产物C1-3的制备:参照实施例1中(IV)的操作,用质量比为1:10的Q5-3与吡啶反应,得到目标光敏剂C1-3(产率:50%)。HR-MS(ESI):[M]+:Calcdfor[C36H40ClN4O]+579.2885;found579.2886。(III)参照实施例1中(V)的操作,证明C1-3溶解度大于5mg/mL;且在350~600nm波长范围有较强吸收峰,见图5。(IV)参照实施例1(VI)的操作,结果同样证明了目标光敏剂C1-3在473nm激光照射下能产生活性氧物质,见图6。实施例13重复实施例12,其不同在于,为环庚酮,催化剂为无水碳酸钾,以(2-吡啶基苯甲酮)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率44%。实施例14重复实施例12,其不同在于,为环辛酮,催化剂为六氢吡啶,以(吖啶)代替吡啶作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率29%。实施例15一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间体化合物S2-1的制备:参照实施例1中(I)的操作,用摩尔量1:2的商业可得的氯苯和vilsmeier 试剂反应,得到中间产物S2-1(产率96%)。(II)中间体化合物Q2-3的制备:参照实施例1中(I)的操作,用摩尔量1:3的商业可得的N,N-二(2-溴乙基)苯胺和vilsmeier试剂反应,得到中间产物Q2-3(产率98%)。(III)中间体化合物S3-1的制备:参照实施例1中(II)的操作,用摩尔量为1:1的S2-1与环己酮反应,催化剂为氢氧化钠,得到中间产物S3-1(产率:50%)。(IV)中间体化合物S4-1的制备:参照实施例1中(III)的操作,用摩尔量1:1的S3-1和Q2-3反应,催化剂为氢氧化钠,得到中间产物S4-1(产率88%)。(V)目标产物C2-1的制备:参照实施例1中(IV)的操作,用质量比为1:30的S4-1与商业可得的三乙胺反应,得到目标光敏剂C2-1(产率:50%)。HR-MS(ESI):[M]+:Calcdfor[C42H69Br2N4O]+803.3833;found803.3847。实施例16重复实施例15,其不同在于,为环戊酮,催化剂为氢氧化钾,以(2-苄基吡啶)代替三乙胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率45%。实施例17重复实施例15,其不同在于,以(吡啶)代替三乙胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率78%。实施例18重复实施例15,其不同在于,以(2,2-联吡啶)代替三乙胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率38%。实施例19重复实施例15,其不同在于,为环庚酮,催化剂为无水碳酸钠,以(喹啉)代替三乙胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率65%。实施例20重复实施例15,其不同在于,为环丁酮,催化剂为无水碳酸钠,以(2-吡啶基苯甲酮)代替三乙胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率67%。实施例21一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间体化合物Q2-4的制备:参照实施例1中(I)的操作,用摩尔量1:2的商业可得的N-(2-溴乙 基)-N-甲基苯胺和vilsmeier试剂反应,得到中间产物Q2-4(产率98%)。(II)中间体化合物S2-2的制备:参照实施例1中(I)的操作,用摩尔量1:2的商业可得的2-(氯乙基)苯和vilsmeier试剂反应,得到中间产物S2-2(产率98%)。(III)中间体化合物S3-2的制备:参照实施例1中(II)的操作,用摩尔量为1:1的Q2-4与环己酮反应,催化剂为六氢吡啶,得到中间产物S3-2(产率:45%)。(IV)中间体化合物S4-2的制备:参照实施例1中(III)的操作,用摩尔量1:1的S2-2和S3-2反应,催化剂为六氢吡啶,得到中间产物S4-2(产率60%)。(V)目标产物C2-2的制备:参照实施例1中(IV)的操作,用质量比为1:40的S4-2与商业可得的三苯胺反应,得到目标光敏剂C2-2(产率:50%)。HR-MS(ESI):[M]+:Calcdfor[C61H57BrN3O]+926.3680;found926.3677。实施例22重复实施例21,其不同在于,为环丁酮,催化剂为氢氧化钾,以(二吡啶基乙烷)代替三苯胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率35%。实施例23重复实施例21,其不同在于,为环戊酮,催化剂为无水碳酸钾,以(2,2-联喹啉)代替三苯胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率57%。实施例24重复实施例21,其不同在于,为环丁酮,催化剂为无水碳酸钠,以(吖啶)代替三苯胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率67%。实施例25重复实施例21,其不同在于,为环庚酮,催化剂为无水碳酸钾,以(7,8-苯并喹啉)代替三苯胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率77%。实施例26重复实施例21,其不同在于,为环戊酮,催化剂为氢氧化钾,以(4,7-二氮杂菲)代替三苯胺作为反应原料,其它条件不变。得到 产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率47%。实施例27重复实施例21,其不同在于,为环辛酮,催化剂为氢氧化钠,以(二苯基-2-吡啶甲烷)代替三苯胺作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率55%。实施例28一种水溶性阳离子亚苄基环烷烃酮光敏剂,其合成方法如下:(I)中间体化合物S2-3的制备:参照实施例1中(I)的操作,用摩尔量1:2的商业可得的2-碘代乙基苯和vilsmeier试剂反应,得到中间产物S2-3(产率96%)。(II)中间体化合物S3-3的制备:参照实施例1中(II)的操作,用摩尔量为1:1的S2-3与环庚酮反应,催化剂为碳酸钾,得到中间产物S3-3(产率:40%)。(III)中间体化合物T1-1的制备:参照实施例1中(III)的操作,用摩尔量1:1的S3-3和S2-1反应,催化剂为碳酸钾,得到中间产物T1-1(产率78%)。(IV)目标产物C3-1的制备:参照实施例1中(IV)的操作,用质量比为1:30的T1-1与商业可得的三苯基膦反应,得到目标光敏剂C3-1(产率:50%)。HR-MS(ESI):[M]+:Calcdfor[C59H52IOP2]+965.2533;found965.2539。实施例29重复实施例28,其不同在于,为环戊酮,催化剂为氢氧化钠,以(7,8-苯并喹啉)代替三苯基膦作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率56%。实施例30重复实施例28,其不同在于,为环丁酮,催化剂为氢氧化钠,以(4,7-二氮杂菲)代替三苯基膦作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率44%。实施例31重复实施例28,其不同在于,为环己酮,催化剂为碳酸钠,以(二苯基-2-吡啶甲烷)代替三苯基膦作为反应原料,其它条件不变。得到产物水溶性阳离子亚苄基环烷烃酮光敏剂,产率28%。实施例32重复实施例28,其不同在于,以(吡啶)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例33重复实施例28,其不同在于,以(联吡啶)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例34重复实施例28,其不同在于,以(喹啉)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例35重复实施例28,其不同在于,以(2-吡啶基苯甲酮)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例36重复实施例28,其不同在于,以(吖啶)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例37重复实施例28,其不同在于,以(2-苄基吡啶)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例38重复实施例28,其不同在于,以(二吡啶基乙烷)代替三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例39重复实施例28,其不同在于,以(2,2-联喹啉)代替 三苯基膦作为反应原料,其它条件不变,得到产物水溶性阳离子亚苄基环烷烃酮光敏剂。实施例40将实施例2中制得的光敏剂用于光动力灭活金黄色葡萄球菌的实验(I)将细菌在肉汤培养基中复苏,通过测定600nm的吸光度值来确定细菌的浓度。(II)利用无菌的96孔板进行最小抑菌浓度(minimuminhibitoryconcentration,MIC)的测定。见示意图7,96孔板中每相邻的四个孔,例如C1,C2,D1和D2所加光敏剂浓度是相同的。按照示意图所示浓度,每孔加入100微升不同光敏剂浓度的肉汤溶液。其中,阴性对照组为B+B组(100微升肉汤中加入10微升的细菌),阳性对照组为GM组(100微升肉汤加入1%的庆大霉素),空白对照组为Broth组(100微升肉汤中加入10微升PBS)。上述96孔板中的细菌浓度约为5×105CFU每毫升。(III)利用532nm的激光(30Jcm-2,10min)照射上述96孔板后,放入细菌培养箱继续培养18小时。然后利用0.0675%的刃天青钠盐溶液进行显色。(IV)经过4小时的显色反应,96孔板中框出浓度所对应的光敏剂浓度为最低抑菌浓度。见图8。实施例41参照实施例40中的操作,将实施例2中制得的光敏剂用于光动力灭活大肠杆菌的实验,见图9。实施例42将实施例2中制得的光敏剂用于光动力灭活白念珠菌的实验。(I)取适量菌液接种到沙氏葡萄糖肉汤培养基中,培养48~72小时。(II)取适合浓度的真菌溶液中加入不同浓度的光敏剂,使光敏剂的最终浓度为25微摩尔、50微摩尔和100微摩尔。摄取1小时后,利用532nm的激光对上述混合液进行光照(30Jcm-2,10min)。并于暗处培养24小时。(III)利用涂布菌板法,观察不同浓度下的菌落数目,测定光敏剂的抗菌活性。见图10。实施例43将实施例8中制得的光敏剂用于光动力灭活金黄色葡萄球菌的实验,见图11。实施例44将实施例8中制得的光敏剂用于光动力灭活大肠杆菌的实验,见图12。实施例45将实施例8中制得的光敏剂用于光动力灭活白念珠菌的实验,见图13。实施例46将实施例13中制得的光敏剂用于观察细胞摄取此类光敏剂的实验。(I)将细胞以105每孔植入玻璃底培养皿中(直径35毫米),24小时后换成含10微摩尔光敏剂的培养液,继续培养4小时;(II)向培养液中加入100纳摩尔荧光探针继续培养20分钟,以PBS清洗去除游离探针;(III)加入1毫升PBS溶液,用荧光共聚焦显微镜观察光敏剂于细胞的定位图。见图14。实施例47将实施例15中制得的光敏剂用于脂水分配比的测定实验将一定量的光敏剂溶于2mLPBS中,然后加入2mL正辛醇,将混合溶液震摇3min,再置于超声波中振荡5min,然后在每分钟5000转的速度下离心5分钟,使两相分离。测定两相中的光敏剂的吸收光谱图,通过Lambert-beer定律计算得到光敏剂在两相中的浓度,脂水分配比(LogPC)即为光敏剂在两相中的浓度比值。见表1。实施例48将实施例21中制得的光敏剂用于脂水分配比的测定实验,见表1。实施例49将实施例28中制得的光敏剂用于脂水分配比的测定实验,见表1。实施例50将实施例31中制得的光敏剂用于光动力灭活烟草花叶病毒。向96孔板中的100μL不同浓度(0.125、0.25、0.5、1.0、2.0、4.0、8.0、16.0、32.0和64.0μM)的光敏剂溶液中加入适量的烟草花叶病毒并保证病毒的终浓度为2x1011pfu/mL,在黑暗中震荡培养30分钟,用波长为550nm左右的Oriel91192太阳能模拟器(5mW/cm2,50min)照射后,将96孔板中的混合溶液转移到含有培养基的琼脂盘上,用菌落计数法计算烟草花叶病毒的成活率。其中,不含光敏剂的同浓度的菌悬液用作对照组。表1光敏剂名称LogPCC2-1-2.66C2-25.72C3-14.58显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1