一种富集循环肿瘤DNA的方法和试剂与流程
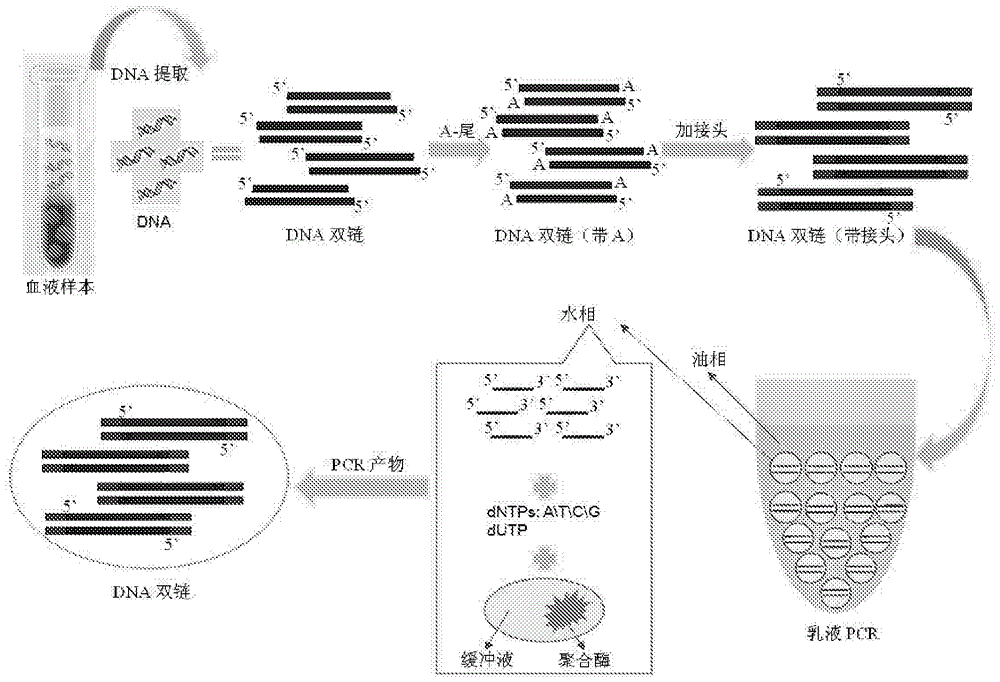
本发明涉及pcr(polymerasechainreaction,聚合酶链式反应)超微量平行扩增
技术领域:
,尤其涉及一种富集循环肿瘤dna的方法和试剂。
背景技术:
:2003年以来,随着人类基因组计划的开展,一代测序(sanger)和二代测序(ngs)技术以其灵敏度和准确性逐步占据分子诊断的市场。分子水平的基因组学也成为了21世纪生命科学领域的研究热点。基因测序技术的趋于临床应用化,各种癌症相关基因的发现,癌症基因组计划的展开,为癌症的早期诊断、个体化治疗,以及预后带来了新的曙光。目前,基因组dna的获得主要来源于组织细胞,包括:活体组织细胞破裂后提取;组织细胞石蜡样本体外pcr扩增。而对于早期的患者,如何从dna水平诊断癌症的发生,甚至预防,这仍然存在技术壁垒。恶性肿瘤的生存检测数据是反映一个地区肿瘤负担以及评价医疗资源和防治水平的重要信息。我国全国肿瘤登记中心的2003-2005年癌症生存率分析表明,在我国癌症5年相对生存率仅为30.9%。主要癌症的生存率分别为:肺癌16.1%,胃癌27.4%,肝癌10.1%,食管癌20.9%,结直肠癌47.2%,乳腺癌73%。其中,城乡癌症生存率差异明显。而在发达国家,结直肠癌和乳腺癌的五年生存率,分别达到60%和85%,预后较差的肝癌和肺癌的五年生存率也高于我国,达到15%-20%(相关数据来自lancet)。可见,要提高我国癌症患者的五年生存率,需从“三早”做起,早发现,早诊断,早治疗。目前,临床应用的癌症诊断和评估手段主要是影像学定位和肿瘤标记物定性相结合。影像学包括:x射线(胸部透视、x线胸片、低剂量螺旋ct)、磁共振(mir)、放射性物质(核骨素扫描)、pet-ct。x射线作为一种常规的筛查方法,主要确定肿瘤的大小和位置,要进行确诊还需结合肿瘤标志物的检测;mir检测肿瘤是否扩散到脑或者脊髓,核骨素扫描则是检测肿瘤是否扩散到骨头,这两种检测技术主要用于肿瘤iii期之后的诊断;pet(positronemissioncomputedtomography),全称正电子发射计算机断层扫描,是目前唯一的用解剖形态方式进行功能、代谢和受体显像的技术,具有无创伤性的特点,pet-ct是pet技术和ct相结合,筛查肿瘤的位置和扩散情况,是临床上用以诊断和指导治疗肿瘤最佳手段之一。但是,这种诊断方法费用较高,且不属于医保范畴,很多患者因无法支付昂贵的检查费而不得不放弃使用。影像学的检测主要用于恶性肿瘤的定位,要定性癌症的类别和分期,还需要对肿瘤标志物进行评估,根据其生化或免疫特性可以识别或诊断肿瘤。肿瘤标记物是由肿瘤细胞产生和释放的某种物质,常以抗原、酶、激素等代谢产物的形式存在于肿瘤细胞内或宿主体液中。用于临床检测的肿瘤标记物有:甲胎蛋白(afp)、癌胚抗原(cea)、糖类抗原家族(ca125、ca15-3等)等。然而,在癌症确诊阶段,以上临床应用的常规影像学所检测到的恶性肿瘤,直径一般在1em以上,肿瘤细胞的数目达到109之多,重量超过1g。pet-ct灵敏度高,可以检测的肿瘤也要达到0.5cm以上(h.lietal.2013;步召德,薛钟麒等,2000)。也就是说,影像学检测到的肿瘤患者,大多数属于中晚期,已经错过了癌症治疗的最佳时期。在癌症早期,即便检测到肿瘤标记物,影像学检测不到肿瘤组织的具体位置,也无法最终确诊肿瘤,而进行及早治疗用药,只能任其发展恶化。肿瘤的耐药性和治愈后的复发是导致肿瘤患者死亡的最重要原因之一,破解这两方面的难题已经成为当今世界肿瘤疾病的研究热点。癌症治疗过程中,目前的临床应用缺乏一种有效的用药评估手段。以肺癌为例,吉非替尼(易瑞沙)、厄洛替尼(特罗凯)是非小细胞肺癌(nsclc)的治疗药物,而临床数据表明并不是所有的非小细胞肺癌患者都适合使用此类药物。进一步研究发现带有egfr(epidermalgrowthfactorreceptor)突变的患者接受易瑞沙治疗反应率比无选择性的患者高40%以上(tonys.mok,m.d.etal.2009;hidat,okamotoi,kashiit,etal.2008;kimurah,kasaharak,kawaishim,etal.2006)。吉西他滨是一种破坏细胞复制的二氟核苷类抗代谢物抗癌药,适用于治疗中、晚期非小细胞肺癌。研究证实:rrm1(rrm1基因定位于1号染色体短臂,编码核糖核苷酸还原酶m1亚单位)表达量低的患者接受吉西他滨治疗疾病控制率高30%以上(leeetal.2010)。欧洲药监局明确规定,非小细胞肺癌患者在使用易瑞沙前必须检测egfr基因,转移性大肠癌患者在使用靶向药物爱必妥和维克替比治疗前必须检测kras基因(来自欧洲药监局官方网站http://www.ema.europa.eu/ema/)。由此可见,对于恶性肿瘤的治疗,用药指导评估是优化治疗方案,实现有效治疗的重要前提和基础。肿瘤发生、发展的系统生物学研究得出结论,dna突变或者由遗传获得的缺陷基因是导致耐药性的关键,因此,从基因水平进行个体化用药指导和评估,大势所趋。除此之外,临床治疗癌症的预后,也存在检测盲期,例如:很多癌症患者通过手术切除肿瘤组织,以防止其继续扩散而危及生命,术后需定期活检复查进行预后评估。因影像学检测辐射大、对人体造成伤害,癌症患者术后多以年为单位进行复查,这大大影响了预后评估的时效性,同时也反映出癌症诊断领域迫切需要一种更加简便、具无创伤性的检测技术。总而言之,目前临床医学对于早期癌症的筛查、诊断、治疗和预后,是癌症治疗的重大突破口,也是提高癌症五年生存率的关键所在。无论是医学研究,还是推广到临床应用,这一领域都存在重大的技术难题。1947年,mandel和metais在血液、滑膜液和脑脊液等体液中,发现了一种胞外dna,主要以dna蛋白质复合物或者游离dna两种形式存在(mandel&metais.1947)。到了80年代,leon等人研究发现,肿瘤患者外周血清dna水平大大高于正常人(leonetal.1977)。且研究发现,在肿瘤患者的血浆和血清中检测到了与原发肿瘤相一致的癌基因突变。这表明,循环dna作为一种新的肿瘤标志物,将在肿瘤的诊断、治疗和预后检测等领域发挥重要作用。循环肿瘤dna(circulatingtumordna,ctdna)是肿瘤细胞体细胞dna经脱落或者当细胞凋亡后释放进入循环系统,可以被定性、定量和追踪。对于这类ctdna的成功捕获,携带信息的准确解读为癌症早期基因信息的获取,癌症的早期诊断、预后检测、耐药评估提供了一条准确的途径。但是,这类技术目前仍未得到有效解决和广泛应用。原因之一在于:ctdna在外周血中的含量非常低,尤其是与正常dna(normaldna,ndna)相比,其相对含量极低,目前的检测技术还难以达到直接检测外周血中ctdna的水平。因此,需要先通过dna富集的方法将目标ctdna富集起来,然后在通过对富集的ctdna进行测序,得到其携带信息,并根据测序得到的携带信息来解读个体的患病情况。技术实现要素:本发明提供一种富集循环肿瘤dna的方法和试剂,该方法能够有效地捕获外周血液血浆中的ctdna,并且进行单分子高保真超微量平行扩增,提供了足够量ctdna以进行后续的测序检测使用。根据本发明的第一方面,本发明提供一种富集循环肿瘤dna的方法,包括如下步骤:单分子平行扩增:将水相和油相混合、振荡配成乳液pcr反应体系并进行乳液pcr扩增,其中所述水相中含有作为模板dna的外周血液血浆dna、正反向引物、dntps、pcr缓冲液和dna聚合酶,所述外周血液血浆dna的两端连有接头序列,所述正反向引物分别与两端的接头序列互补配对;分离水相和油相:所述乳液pcr扩增后,分离水相和油相,得到水相的pcr扩增产物;捕获循环肿瘤dna:使用特异性结合循环肿瘤dna的探针序列捕获所述水相的pcr扩增产物中的循环肿瘤dna。作为本发明的优选方案,所述乳液pcr反应体系的水相中还含有dutp;优选地,所述dutp与所述dntps的摩尔比为1∶1000~1∶10,更优选为1∶100。作为本发明的优选方案,所述接头序列为测序设备中使用的测序接头序列;优选地,所述测序设备为illumina测序仪,所述测序接头序列对应的正反向引物序列分别为5’-tccctacacgacgctcttccgatct-3’(seqidno:1)和5’-tgaacctgaaccgctcttccgatct-3’(seqidno:2);优选地,所述测序设备为lifetech测序仪,所述测序接头序列对应的正反向引物序列分别为5’-ccatctcatccctgcgtgtctccga-3’(seqidno:3)和5’-ccgctttcctctctatgggcagtcg-3’(seqidno:4)。作为本发明的优选方案,所述水相与油相的体积比为1∶10~1∶1,优选为1∶5~1∶2。作为本发明的优选方案,所述dna聚合酶为高保真dna聚合酶,优选为高保真的klenowfragment(dna聚合酶i,dnapolymerasei)、kapahifi家族高保真dna聚合酶、phusion家族高保真dna聚合酶或q5家族高保真dna聚合酶。作为本发明的优选方案,所述水相中外周血液血浆dna总量1-100ng、正反向引物终浓度为0.1-1μm、dntps终浓度为0.5-2mm、pcr缓冲液终浓度为1倍、dna聚合酶终浓度为0.1-1u;优选地,所述水相中外周血液血浆dna总量1-10ng、正反向引物终浓度为0.5μm、dntps终浓度为1mm、pcr缓冲液终浓度为1倍、dna聚合酶终浓度为0.25u;优选地,所述乳液pcr扩增的程序为93-95℃1-3min;93-95℃5-20s,61-65℃10-20s,71-73℃5-20s,40-60个循环;71-73℃4-10min;优选地,所述乳液pcr扩增的程序为94℃2min;94℃10s,63℃15s,72℃10s,50个循环;72℃5min。作为本发明的优选方案,所述捕获循环肿瘤dna中使用的特异性结合循环肿瘤dna的探针序列带有生物素修饰;所述探针序列与所述循环肿瘤dna特异性结合以后,通过链霉亲和素磁珠特异性结合生物素而捕获所述循环肿瘤dna;需要说明的是:生物素修饰可以在探针序列的任意位置,比如探针序列的5’端、3’端或中间任意碱基位置。优选地,在使用所述探针序列捕获所述循环肿瘤dna过程中,使用封闭序列封闭所述接头序列,其中所述封闭序列特异性结合外周血液血浆dna两端的接头序列。作为本发明的优选方案,所述捕获循环肿瘤dna,具体通过使用特异性结合循环肿瘤dna的探针序列作为引物,进行pcr扩增而实现,其中所述作为引物的探针序列为特异性结合循环肿瘤dna的序列。根据本发明的第二方面,本发明提供一种富集循环肿瘤dna的试剂,所述试剂包括如下组成部分:乳液pcr扩增组分,包括水相和油相,其中所述水相包含正反向引物、dntps、pcr缓冲液和dna聚合酶,所述正反向引物分别与连接于外周血液血浆dna两端的接头序列互补配对;捕获循环肿瘤dna组分,包括特异性结合循环肿瘤dna的探针序列,用于捕获所述水相的pcr扩增产物中的循环肿瘤dna;优选地,所述乳液pcr扩增组分的水相中还含有dutp;优选地,所述dutp与所述dntps的摩尔比为1∶1000~1∶10,更优选为1∶100;优选地,所述dna聚合酶为高保真dna聚合酶,优选为高保真的klenowfragment(dnapolymerasei)、kapahifi家族高保真dna聚合酶、phusion家族高保真dna聚合酶或q5家族高保真dna聚合酶。作为本发明的优选方案,所述探针序列带有生物素修饰,并且所述试剂还包括链霉亲和素磁珠,所述探针序列与所述循环肿瘤dna特异性结合以后,通过链霉亲和素磁珠特异性结合生物素而捕获所述循环肿瘤dna;优选地,所述试剂还包括封闭序列,所述封闭序列特异性结合外周血液血浆dna两端的接头序列,用于在使用所述探针序列捕获所述循环肿瘤dna过程中封闭所述接头序列;优选地,所述探针序列为特异性结合循环肿瘤dna的序列,用于进行pcr扩增得到富集的循环肿瘤dna。本发明的方法通过乳液pcr与探针捕获技术的结合,实现单分子高保真超微量平行扩增以及有效地捕获外周血液血浆中的ctdna,提供了足够量ctdna以进行后续的测序检测使用。与传统的病理学检测方法相比,本发明的方法无需组织细胞活检取样,只要少量外周血液血浆即可大量富集ctdna,用于后续的测序检测,因此实现了无创检测。不仅节省了检测费用、提高了诊断准确性,而且打破了癌症的检测极限,由传统的中晚期检测,提前到早期诊断,并且本发明的方法操作简便。需要说明的是:本发明的富集循环肿瘤dna的方法本身还不能确定患者的患癌情况,因为该方法本身只在于富集外周血液血浆中的循环肿瘤dna,为后续测序分析携带信息准备足够的样本材料,因此富集循环肿瘤dna的方法本身并不能作为诊断方法。附图说明图1为本发明中的乳液pcr的原理和过程示意图;图2为本发明中的生物素-寡核苷酸捕获ctdna的原理和过程示意图;图3为本发明实施例1捕获得到的ctdna凝胶电泳检测结果;图4为本发明中的引物pcr捕获ctdna的原理和过程示意图;图5为本发明实施例2捕获得到的ctdna凝胶电泳检测结果;图6为本发明实施例3捕获得到的ctdna凝胶电泳检测结果。具体实施方式下面通过具体实施例对本发明作进一步详细说明。除非特别说明,下面实施例中所使用的技术均为本领域内的技术人员已知的常规技术;所使用的仪器设备和试剂等,均为本领域内的技术人员可以通过公共途径如商购等获得的。本发明主要解决了以下两个问题:1、ctdna和ndna的平行扩增问题:由于ctdna与ndna的比例约为1/1000,如何有效地平行扩增是决定评判结果的关键。传统的pcr扩增方法的结果是ndna随扩增进行所占比例越来越大,而ctdna随扩增进行所占比例越来越小。本发明的方法,对外周血液血浆中的dna(包含ctdna和ndna)进行超微量平行扩增,确保扩增后,两者比例仍为1/1000。2、ctdna的捕获问题:对于上面平行扩增的dna中包含ctdna和ndna,需要从全基因组中捕获出癌症基因部分,得到ctdna,本发明的方法通过捕获ctdna的探针序列成功实现ctdna捕获富集。本发明的方法能够有效捕获ctdna,进行单分子高保真扩增,为后续检测提供足够量的ctdna。本发明的方法主要包括两部分,分别针对上述两个问题提出了解决方案。第一部分,是针对第一个问题的解决方案:用乳液pcr(emulsionpcr,epcr)单分子平行扩增ctdna和ndna。第二部分,是针对第二个问题的解决方案:用生物素标记的寡核苷酸(biotin-oligo)杂交癌症基因,然后结合链霉亲和素磁珠(streptavidinmagneticbeads),捕获分离出癌症基因;或者设计引物,pcr扩增ctdna,捕获分离出癌症基因。本发明分两部分列出具体技术方案,如下:第一部分:乳液pcr请参考图1,乳液pcr(emulsionpcr,epcr)是利用油包水乳液体系中的微液滴作为反应器进行pcr扩增。epcr体系包括油相和水相的反应体系。油相作为载体,本发明中使用的油相可以不作限定,优选可以采用表1所示的油相体系。表1本发明优选采用的油相配方体系水相中反应体系中包括:dna模板(dnatemplate)、dntps(包括datp、dttp、dctp、dgtp)、pcr缓冲液、dna聚合酶(dnapolymerase)和双蒸水(ddh2o)。其中,dna模板选取外周血液血浆中的dna,其中包含双链的ctdna和ndna。在进行epcr之前,dna双链进行三步修饰,第一步,对双链dna末端进行修复;第二步将“a”加入到dna片段的3’末端;第三步,在dna末端加上特定的接头(adapter),用于后续的测序,接头的选取需结合具体的测序设备(常用的测序仪器如illumina,lifetech等)。研究发现,在水相体系中加入少量dutp,有利于反应进行,其中dutp与dntps的摩尔比为1∶1000~1∶10较佳,更优选为1∶100。选用illumina系统测序仪时,接头引物部分序列如下:正向引物(5’-3’):tccctacacgacgctcttccgatct(seqidno:1);反向引物(5’-3’):tgaacctgaaccgctcttccgatct(seqidno:2)。选用lifetech系统测序仪时,接头引物部分序列如下:ion_a(red,5’-3’):ccatctcatccctgcgtgtctccga(seqidno:3);ion_p1(blue,5’-3’):ccgctttcctctctatgggcagtcg(seqidno:4)。除此之外,水相反应体系中的其他组分dntps、dutp、缓冲液(buffer)、dna聚合酶可以提前配置为mastermix,见表2,分装后置于-20℃冻存待用。表2mastermix配置比例(总体积为200μl)试剂量终浓度(/μl)10×buffer(含有mg2+)20μl1×phi29polymerase(10u/μl)5μl0.25udntps(10mm)20μl1.0mmdutp(0.1mm)20μl10μmddh2o135μl-epcr中,水相/油相可选取不同的比例,如:75μl/400μl;240μl/960μl;200μl/400μl;300μl/400μl等。以水相/油相比例200μl/400μl为例,油相选取配方2,400μl油相中,加入160μl硅油ar20、120μl环甲基硅氧烷(及)硅酮多元醇共聚物(5225cformulationaid)、120μl749fluid。水相中epcr体系及循环参数如表3和表4。表3pcr体系各组分比例表4pcr循环参数水相和油相依次加入1.5ml不沾壁(non-stick)的反应管中。两相混合后,将装有反应体系的反应管放在组织破碎仪(qiagentissuelyserii)中震荡90秒,参数设为13hz。取下反应试管,将油水体系分装至pcr反应管,根据表4的设置参数进行pcr反应。接下来,pcr结束后,分离pcr反应管中的水相和油相。本发明可以采用三种分离方法,方法一:去掉反应管上部的油相,向反应管中加入400μl石蜡油,涡旋或者用移液管反复吸取管中溶液,直至水相的乳球破裂,水相和油相充分混合。将混合均匀的液体从pcr反应管转移到新的不沾壁反应管中,用400μl硅油(siliconoil)洗脱pcr反应管,洗脱液加入上述水相/油相混合液,充分涡旋,15000rpm离心2min,去掉上层油相。接下来需要对混合液进行洗脱。硅油洗脱三次,加入500μl硅油,充分涡旋,15000rpm离心1min弃上层油相,再重复2次操作。分离的到水相的pcr反应产物。方法二:收集反应管中的水相和油相,9000g离心5min,去掉油相,此时水相仍然呈乳球状沉淀在反应管底部,加入400μl(两倍体积)的饱和乙醚,混合物充分涡旋,15000rpm离心2min,去掉上层的乙醚;乙醚重复洗涤一次,得到水相的pcr反应产物。方法三:收集反应管中的水相和油相,15000rpm离心10min弃上层油相,加入400μl(两倍体积)的用苯酚/三氯甲烷,混合物充分涡旋,15000rpm离心2min,去掉上层的苯酚/三氯甲烷;苯酚/三氯甲烷重复洗涤一次,得到水相的pcr反应产物。pcr聚合酶,一般选用高保真的klenowfragment(dnapolymerasei)、kapahifi家族、高保真的phusion家族以及高保真的q5家族dna聚合酶等。需要注意的是,得到的dna产物是两端含有接头的单链dna,为避免在捕获ctdna时,dna末端的接头发生互补而引入ndna,得到的epcr产物在进行后续实验时,加入与接头互补的寡核苷酸序列,即:最后得到的dna单链,在末端是双链接头形式。且体系中有dna聚合酶和相应的缓冲液,在进行下一步实验时,考虑是否需要将酶失活(一般65-70℃,10min即可,具体温度根据使用的酶而定)。第二部分:第一部分得到单链dna,在此要捕获出ctdna,有两种方案,分别列出如下。方案一:请参考图2,主要是通过磁珠特异性的生物素标记的寡核苷酸(biotin-oligo),捕获出单链ctdna。此方案过程主要是:结合-洗涤-洗脱。生物素标记的寡核苷酸,包括两部分:基因互补配对部分和生物素标记部分。生物素可以特异性吸附在链霉亲和素磁珠表面,通过分离磁珠,洗涤去掉其它组分并洗脱,即可分离出ctdna;基因互补配对部分在捕获ctdna的过程中与ctdna进行碱基互补配对结合。为提高捕获ctdna效率,基因互补配对部分的长短可根据具体情况调整,一般50bp左右。在超微量平行扩增的dna产物中,加入设计好的生物素标记的寡核苷酸,98℃放置5min,60℃震荡2小时,然后冷却至室温。加入混匀的洗净的链霉亲和素磁珠,室温下震荡30min。磁珠必须在液面以下,确保磁珠与含有生物素标记的ctdna-生物素-寡核苷酸(ctdna-biotin-oligo)复合物充分结合。接下来,对磁珠进行洗脱。首先,置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复两次此操作,直至液体去除干净;加入300μl1×b&w缓冲液洗涤,快速涡旋,置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复一次此操作;加入300μl1×te缓冲液洗涤,快速涡旋,置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复一次此操作;最后,加入300μl0.125m的naoh溶液,涡旋1sec,在室温中放置10min,然后充分涡旋置于磁力架放置2min,磁珠紧贴在管壁上,取清液,即得到捕获出的ctdna。磁珠用1×te缓冲液洗脱三次后,可重复使用。具体操作如下:第一次洗脱,加入300μl1×te缓冲液,涡旋后快速离心,置于磁力架放置30sec,磁珠紧贴在管壁上,去掉上清;第二次洗脱,加入300μl1×te缓冲液(含0.01%trition)涡旋后快速离心,置于磁力架放置30sec,磁珠紧贴在管壁上,去掉上清;第三次洗脱,加入300μl1×te缓冲液(含0.01%trition),涡旋后快速离心,置于磁力架放置30sec,磁珠紧贴在管壁上,去掉上清;最后,磁珠用50μl1×te缓冲液(含0.01%trition)悬浮,即可。实施例1:肺癌患者血样储存在促凝采血管中,取上清1ml,置于1.5ml的ep管中,13000rpm,离心1min,弃沉淀,上清备用。取上清液的血清400μl,提取dna。采用过柱回收试剂盒进行(tiangen的血清/血浆游离dna提取试剂盒),具体操作:1,加入40μlproteinasek溶液,充分斡旋混匀;2,加入400μlgb缓冲液,2μlcarrierrna(1ng/μl),混匀,58℃温浴10分钟;3,加入400μlethylalcohol,置于室温放置10分钟;4,cr2柱放在2ml的离心管中,将上述溶液转移至cr2柱,12000rpm离心30sec,倒掉管中的废液;5,加入1000μlgd缓冲液,12000rpm离心30sec,弃废液;6,加入1200μlgd缓冲液,12000rpm离心30sec,弃废液;7,重复第6步操作;8,将cr2置于离心机,12000rpm离心2min,室温下放置5min;8,40μltb缓冲液洗脱cr2,室温下放置5min,然后12000rpm离心2min,为充分洗脱,此步可重复操作一遍。提取的血清血浆dna测浓度为6.74ng/μl。提取的血清血浆dna进行epcr,水相/油相比例为200μl/400μl。油相根据表1的配方3进行制备,1ml油相中,700μl乳化剂abilwe90、200μl石蜡油,730μl碳酸二乙基己酯。水相体系中的组分dntps、dutp、缓冲液(buffer)、dna聚合酶根据表2配置为mastermix,其中10x缓冲液20μl,phi29dna聚合酶(10u/μl)5μl,dntps(10mm)20μl,dutp(0.1mm)20μl,灭菌ddh2o134μl,共199μl。dna模版加1μl,因此,水相共200μl。水相200μl和油相400μl,依次加入1.5ml不沾壁的反应管中。两相混合,反应管放在组织破碎仪(qiagentissuelyserii)中震荡90秒,参数设为13hz。然后将600μl反应体系分装于6个反应管中,100μl/管,进行epcr。pcr参数设置三步操作,第一步,预变性,94℃,2min;第二步循环操作,变性,94℃,10sec,引物退火,63℃,15sec,延伸72℃,10sec,共进行35个循环;第三步,终延伸,72℃,5min。得到的epcr反应产物置于4℃保存备用即可。得到epcr的产物进行后期处理,分离水相和油相。具体操作:1,去掉反应管上部的油相,向反应管中加入400μl石蜡油,涡旋或者用移液管反复吸取管中溶液,直至水相的乳球破裂,水相和油相充分混合。2,洗脱反应管,将混合均匀的液体转移到新的不沾壁反应管中,用400μl硅油洗脱pcr反应管,洗脱液加入上述水相/油相混合液,充分涡旋,15000rpm离心2min,去掉上层油相。3,洗脱水相/油相混合液。硅油洗脱三次,加入500μl硅油,充分涡旋,15000rpm离心1min弃上层油相,再重复2次操作。用移液枪将水相的液体,轻轻吸出,转移至新的ep管中,得到水相的pcr反应产物200μl。epcr扩增的dna产物中,含有dna聚合酶,因此,在进行下一步实验之前,要进行dna聚合酶失活,将反应管于65℃,温浴10min。从epcr得到的dna产物,捕获ctdna,设计生物素标记的寡核苷酸(biotin-oligo)。待捕获的egfr第17个外显子(exon17th)序列如下(5’-3’):设计基因互补配对部分(与下划线部分配对结合):5’-/bio/gaggccgatccccagggccaccaccagcagcaagaggagggcccccacca-3’(seqidno:6),其中5’-端部分带有生物素标记。在epcr的dna产物中,加入5’-端部分带有生物素标记的寡核苷酸,捕获ctdna。具体操作:1,200μl水相的pcr反应产物,加入生物素标记的寡核苷酸20μl(寡核苷酸浓度10μm,终浓度1μm),98℃放置5min,60℃震荡2小时,然后冷却至室温。2,加入50μl混匀的洗净的链霉亲和素磁珠,室温下震荡30min。磁珠必须在液面以下,确保磁珠与含有生物素标记的ctdna-生物素-寡核苷酸(ctdna-biotin-oligo)复合物充分结合。3,吸附磁珠。将反应管置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复两次此操作,直至液体去除干净。4,洗脱磁珠。加入300μl1×b&w缓冲液洗涤,快速涡旋,置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复一次此操作;加入300μl1×te缓冲液洗涤,快速涡旋,置于磁力架放置2min,磁珠紧贴在管壁上,去掉液体,再重复一次此操作;4,加入300μl0.125m的naoh溶液,涡旋1sec,在室温中放置10min,然后充分涡旋置于磁力架放置2min,磁珠紧贴在管壁上,取清液,即得到捕获出的ctdna。捕获的dna产物,大小为142bp,取5μl捕获的dna溶液,加入1μl6x上样缓冲液(loadingbuffer),充分混匀,点样进行电泳检测,dnamarker选用100bpplus(genstar),上样1μl。琼脂糖凝胶配置的浓度为2%,取琼脂糖2g,加入100ml1xtae缓冲液中,微波加热2min,至琼脂糖完全溶解,待溶液冷却至50-60℃,加入5μl核酸染料(goldview),倒胶。电泳检测结果如图3所示。其中,捕获的dna溶液上样至第11和第12泳道,根据电泳图分析,目的条带大小符合,即该方法成功捕获得到ctdna,可用于后续其他检测。方案二:请参考图4,主要是通过引物pcr扩增获得ctdna。上述超微量平行扩增的dna产物中,含有dna聚合酶,因此,在进行方案二时要进行dna聚合酶失活,65℃,10min处理,即可。设计pcr引物,包括两部分:基因互补配对部分和接头(adapter)部分。所述pcr引物无需末端修饰,基因互补配对部分位于3’端,pcr扩增时与单链ctdna发生碱基配对;接头部分位于5’端,在后续的基因测序中发挥作用。为提高捕获ctdna效率,基因互补配对部分的长短可根据具体情况调整,一般25bp左右,不低于20bp;在接头部分设计时,结合具体的测序设备选择序列,长度一般不低于20bp。pcr的合成酶,一般选用高保真的klenowfragment(dnapolymerasei)、kapahifi家族、高保真的phusion家族以及高保真的q5家族dna聚合酶等。因此,在设计引物时,引物退火温度应当符合所选合成酶的活性温度。实施例2:实施例1中,epcr扩增的dna产物中,含有dna聚合酶,因此,在进行下一步实验之前,要进行dna聚合酶失活,将反应管于65℃,温浴10min。epcr扩增得到200μldna产物,采用引物pcr捕获ctdna。设计pcr引物,待捕获的egfr第17个外显子(exon17th)序列如下(5’-3’):基因互补配对部分(下划线部分):正向引物:(5’-3’)gcctaagatcccgtccatcgcca(seqidno:7);反向引物:(5’-3’)ctccctctcctgcagcagcctc(seqidno:8)。接头部分(选用illumina系统测序仪):正向引物:(5’-3’)tccctacacgacgctcttccgatct(seqidno:1);反向引物:(5’-3’)tgaacctgaaccgctcttccgatct(seqidno:2)。因此,设计的引物如下,tm表示退火温度(盐浓度为50mm):正向引物:tccctacacgacgctcttccgatctgcctaagatcccgtccatcgcca(seqidno:9,tm:69.7℃);反向引物:tgaacctgaaccgctcttccgatctctccctctcctgcagcagcctc(seqidno:10,tm:68.5℃)。epcr得到的dna产物200μl,测浓度为50ng/μl,取1μl稀释10倍,得到浓度为5ng/μl的dna溶液。以此浓度的dna溶液为模板进行pcr扩增。pcr扩增体系和循环参数如下表4和表5所示。合成酶为kapa2grobusthotstart。表4pcr体系各组分比例表5pcr循环参数pcr产物,大小为192bp(含有引物两端的adapter),取5μlpcr反应产物,加上1μl6x上样缓冲液(loadingbuffer),充分混匀,点样进行电泳检测,dnamarker选用100bpplus(genstar),上样1μl。琼脂糖凝胶的浓度为2%,配置同于实施例1。电泳检测结果如图5所示。其中,捕获的dna溶液上样至第9泳道,根据电泳图分析,目的条带大小符合,即该方法成功捕获得到ctdna。为进一步检测确定,该pcr获得的dna是egfr第17个外显子,对pcr产物进行一代测序。首先,扩大体系进行核酸胶电泳,琼脂糖凝胶配置的浓度为2%,进行胶回收。剩余的95μlpcr反应产物,加上19μl6x上样缓冲液(loadingbuffer),充分混匀,点样进行电泳检测,dnamarker选用100bpplus(genstar),上样6μl。琼脂糖凝胶的制备方法同上。然后,对上述pcr产物进行胶回收(omega胶回收试剂盒),具体操作:1,紫外下切下含有目的条带egfr基因exon17的核酸胶,称重0.50000g(空白ep管重1.04100g,加入切割的胶体重1.54100g);2,加入bindingbufferxp2500μl,55-65℃温浴7min,直至核酸胶完全溶解,混匀;3,将上述溶液转移至干净的离心柱中,离心柱置于新的2ml离心管中,10000g离心1min,弃废液;4,离心柱中加入300μl的bindingbufferxp2,10000g离心1min,弃废液;5,加入700μl的washbufferspw,13000g离心2min,弃废液;6,重复步骤5;7,离心柱放回离心管中,13000g离心2min,弃废液;8,离心柱在室温下放置约5min,直至干燥;9,离心柱放入干净的ep管中,30μl灭菌的ddh2o,室温下放置1min,13000g离心1min,为充分洗脱,此步可重复操作一遍。最后,将回收产物(30ng/μl,≥10μl)送至生工进行一代测序,正向引物测序。从序列比对的结果分析,pcr产物序列与egfr基因的exton17序列比对正确。但是,从测序结果的碱基峰图看出,1-50bpdna碱基的峰值出现套封。血清提取的dna中包含ndna和ctdna,二者比例约为1000∶1,因此出现套峰,表示该部分dna含有ndna和ctdna,ctdna序列可能存在突变现象。实施例3:(选用lifetech系统测序仪):实施例1中,epcr扩增的dna产物中,含有dna聚合酶,因此,在进行下一步实验之前,要进行dna聚合酶失活,将反应管于65℃,温浴10min。epcr扩增得到的dna产物,采用引物pcr捕获ctdna。设计pcr引物,待捕获的egfr第17个外显子(exon17th)序列如下(5’-3’):基因互补配对部分(下划线部分):正向引物:(5’-3’)gcctaagatcccgtccatcgcca(seqidno:7);反向引物:(5’-3’)ctccctctcctgcagcagcctc(seqidno:8)。接头引物部分(选用lifetech系统测序仪):ion_a(red,5’-3’):ccatctcatccctgcgtgtctccga(seqidno:3);ion_p1(blue,5’-3’):ccgctttcctctctatgggcagtcg(seqidno:4)。因此,设计的引物如下:正向引物(5’-3’):反向引物(5’-3’):epcr得到的dna产物200μl,测浓度为50ng/μl,取1μl稀释10倍,得到浓度为5ng/μl的dna溶液。以此浓度的dna溶液为模板进行pcr扩增。pcr扩增体系和循环参数如下表6和表7所示。合成酶为phusiondna聚合酶(phusiondnapolymerase)。表6pcr体系各组分比例表7pcr循环参数pcr产物,大小为192bp(含有引物两端的adapter),取5μlpcr反应产物,加上1μl6x上样缓冲液(loadingbuffer),充分混匀,点样进行电泳检测,dnamarker选用100bpplus(genstar),上样1μl。琼脂糖凝胶的浓度为2%,制备方法同于实施例1。电泳检测结果如下图6所示。其中,捕获的dna溶液上样至第10泳道,根据电泳图分析,目的条带大小符合,即,该方法成功捕获得到ctdna。以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属
技术领域:
的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种检测哺乳动物血液中微量循环肿瘤细胞的特异性方法与制造工艺
- 一种循环肿瘤细胞阴性富集方法与制造工艺
- 高通量测序检测人循环肿瘤DNA EGFR基因的捕获探针及试剂盒的制造方法与工艺
- 用于肿瘤早期筛查的肿瘤血小板RNA定量检测模型及方法与制造工艺
- 肿瘤循环DNA技术检测‑癌症早期易发风险评估数据法的制造方法与工艺
- 一种偶联抗体的仿生免疫磁球及其制备方法与制造工艺
- 一种循环肿瘤细胞快速检测试剂盒及其制备和应用方法与制造工艺
- 循环肿瘤细胞表面标志分子磷脂酰肌醇蛋白聚糖‑3的检测方法与制造工艺
- 使用组合的核酸酶、连接酶、聚合酶和测序反应识别和枚举核酸序列、表达、拷贝或DNA甲基化变化的方法与制造工艺
- 一种循环肿瘤细胞分析仪的制造方法与工艺