一种TLR7激动剂的三氟乙酸盐、晶型B及其制备方法、药物组合物和用途与流程
本发明属于药物化学领域,具体而言,本发明涉及一种tlr7激动剂的三氟乙酸盐、其制备方法、包含该盐的药物组合物以及该盐的医药用途,本发明还涉及上述盐的晶型b、其制备方法、包含该晶型b的结晶组合物、包含该晶型b或其结晶组合物的药物组合物以及它们的医药用途。
背景技术:
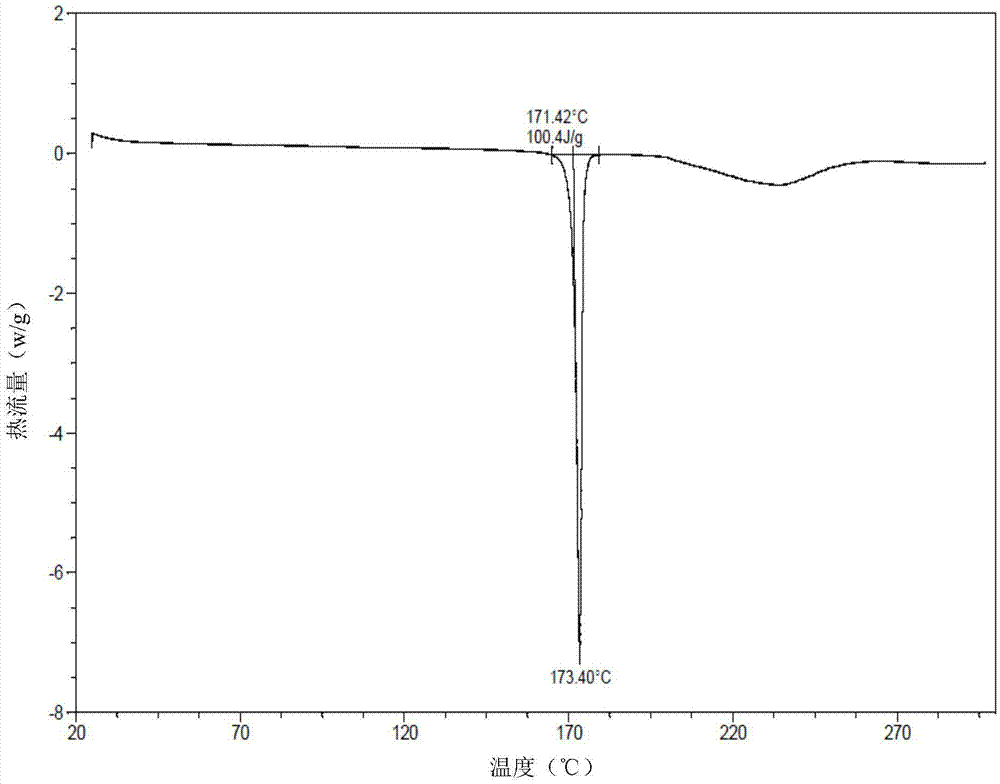
:toll样受体表达于多种免疫细胞。toll样受体识别高度保守结构基序:由微生物病原体表达的病原体相关的微生物模式(pamp)或由坏死细胞释放的损伤相关分子模式(damp)。通过相应的病原体相关的微生物模式(pamp)或损伤相关分子模式(damp)刺激toll样受体引发信号级联导致转录因子如ap-1、nf-κb和干扰素调节因子(脉冲响应函数)的激活。这导致多种细胞反应,包括生产干扰素,促炎性细胞因子和效应细胞因子,从而产生免疫应答。迄今为止哺乳动物中有13种toll样受体已被发现。toll样受体1、2、4、5和6主要表达在细胞表面上,toll样受体3、7、8和9表达在内体中。不同的toll样受体识别不同病原体衍生的配体。对于toll样受体7(tlr7),它主要是由浆细胞样树突细胞(pdc)表达和配体识别而诱导干扰素α(ifn-α)的分泌。toll样受体7(tlr7)和toll样受体8(tlr8)高度同源。因此tlr7配体,在大多数情况下也是tlr8配体。tlr8刺激主要诱导产生细胞因子如肿瘤坏死因子α(tnf-α)和趋化因子。干扰素α是治疗慢性乙型肝炎或丙型肝炎的主要药物之一,而tnf-α是一种促炎细胞因子,过多分泌可能导致严重的副作用。所以对tlr7和tlr8的选择性对于开发tlr7激动剂用于治疗病毒感染性疾病至关重要。tlr7激动剂已有报道,如咪喹莫特,瑞喹莫德,gs-9620。但具备更好的选择性、活性和安全性的新的tlr7激动剂仍然有很大需求。一种这样的小分子为2-丁氧基-7-(4-(吡咯烷-1-基甲基)苄基)-5h-吡咯并[3,2-d]嘧啶-4-胺,它具有以下结构:中国专利申请201410405136.0公开了式i化合物及其合成方法等,在此全文引入作为参考。除了治疗效力外,药物研发者试图提供具有作为药物的性质(所述性质涉及加工、制造、储存稳定性等)的活性分子的适合形式。因此,发现具有所需性质的形式对药物研发至关重要。技术实现要素:一方面,本发明提供了式i所示化合物的三氟乙酸盐:本发明进一步提供了一种药物组合物,该药物组合物中包含有效量的上述盐。本发明进一步提供了上述盐在制备治疗toll样受体7(tlr7)相关疾病中的用途。另一方面,本发明提供了上述盐的晶型b,其中,在x-射线粉末衍射(xrpd)图谱中,具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、14.0°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、22.2°±0.2°、23.5°±0.2°的衍射峰。本发明还提供了上述晶型b的制备方法,该方法包括以下步骤:1)使式i所示化合物的三氟乙酸盐溶于结晶溶剂中,加热溶解;2)冷却析晶、洗涤、干燥。本发明进一步提供了包含晶型b的结晶组合物。在本发明的部分实施方式中,晶型b占结晶组合物重量的50%以上。本发明进一步提供了一种药物组合物,该药物组合物中包含有效量的晶型b或其结晶组合物,此外,该药物组合物还可以含有或不含有药学上可接受的载体、赋形剂和/或介质。本发明更进一步提供了晶型b、其结晶组合物或者所述药物组合物在制备治疗toll样受体7(tlr7)相关疾病的药物中的用途。可通过如下段落[1]至段落[20]中所述的内容对本发明的技术方案加以说明:[1]一种式i所示化合物的三氟乙酸盐,[2]如段落[1]所述的式i所示化合物的三氟乙酸盐,其特征在于,式i所示化合物与三氟乙酸的摩尔比为1:0.5~2,优选1:1。[3]一种药物组合物,所述药物组合物包括有效量的如段落[1]或[2]所述的式i所示化合物的三氟乙酸盐。[4]如段落[1]或[2]所述的式i所示化合物的三氟乙酸盐、或者段落[3]所述的药物组合物在制备治疗toll样受体7相关疾病的药物中的用途。[5]如段落[4]所述的用途,其特征在于,所述疾病为病毒感染。[6]一种式i所示化合物的三氟乙酸盐的晶型b,其特征在于,在x-射线粉末衍射图谱中,具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、14.0°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、22.2°±0.2°、23.5°±0.2°的衍射峰。[7]如段落[6]所述的晶型b,其特征在于,在x-射线粉末衍射图谱中,具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、12.4°±0.2°、14.0°±0.2°、14.9°±0.2°、16.1°±0.2°、16.6°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、21.1°±0.2°、21.8°±0.2°、22.2°±0.2°、23.5°±0.2°的衍射峰。[8]如段落[7]所述的晶型b,其特征在于,在x-射线粉末衍射图谱中,具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、12.4°±0.2°、13.3°±0.2°、14.0°±0.2°、14.9°±0.2°、16.1°±0.2°、16.6°±0.2°、17.3°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、21.1°±0.2°、21.8°±0.2°、22.2°±0.2°、23.1°±0.2°、23.5°±0.2°、24.4°±0.2°、24.7°±0.2°、26.1°±0.2°、28.2°±0.2°的衍射峰。[9]如段落[6]-[8]中任一项所述的晶型b,其特征在于,所述晶型b具有基本上如图1所示的x-射线粉末衍射图。[10]如段落[6]-[9]中任一项所述的晶型b,其特征在于,用dsc进行表征时,所述晶型b的起始温度为171.4℃±5℃,峰值温度为173.4℃±5℃。[11]如段落[6]-[10]中任一段所述的晶型b的制备方法,所述方法包括以下步骤:1)使式i所示化合物的三氟乙酸盐溶于结晶溶剂中,加热溶解;2)冷却析晶、洗涤、干燥。[12]如段落[11]所述的制备方法,其特征在于,步骤1)中所述结晶溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、丙酮、乙酸乙酯或它们混合溶剂、或者水。[13]如段落[12]所述的制备方法,其特征在于,所述结晶溶剂为乙醇。[14]如段落[11]-[13]中任一项所述的制备方法,其特征在于,步骤1)中,每1g式i所示化合物的三氟乙酸盐对应加入的结晶溶剂量为1~5ml。[15]如段落[14]所述的制备方法,其特征在于,每1g式i所示化合物的三氟乙酸盐对应加入的结晶溶剂量为2~3ml。[16]如段落[11]-[16]中任一项所述的制备方法,其特征在于,步骤1)中,所述加热溶解是使式i所示化合物的三氟乙酸盐与结晶溶剂形成均相体系。[17]一种结晶组合物,其特征在于,段落[6]-[10]中任一段所述的晶型b占结晶组合物重量的50%以上,优选80%以上,更优选90%以上,最优选95%以上。[18]一种药物组合物,包含有效量的如段落[6]-[10]中任一段中所述晶型b或者如段落[21]所述的结晶组合物。[19]如段落[6]-[10]中任一段所述的晶型b、如段落[17]所述的结晶组合物、或者如段落[18]所述的药物组合物在制备治疗toll样受体7相关疾病的药物中的用途。[20]如段落[19]所述的用途,其特征在于,所述疾病为病毒感染。附图说明图1式i所示化合物的三氟乙酸盐的晶型b的xrpd谱图。图2式i所示化合物的三氟乙酸盐的晶型b的dsc谱图。具体实施方式本发明采用下述缩略词:sem-cl代表2-(三甲硅烷基)乙氧甲基氯;dipea代表二异丙基乙基胺;tfa代表三氟乙酸;dmf代表n,n-二甲基甲酰胺;n-buoh代表正丁醇;nh3·h2o代表氨水;na代表金属钠;xrpd代表x射线粉末衍射;dsc代表差热分析。本发明提供了式i所示化合物的三氟乙酸盐:在本发明的部分实施方式中,本发明所述的式i所示化合物的三氟乙酸盐中,式i所示化合物与三氟乙酸的摩尔比为1:0.5-2,优选1:1。本发明所述式i所示化合物的三氟乙酸盐可以采用常规的三氟乙酸盐制备工艺来制备。本发明还提供了式i所示化合物的三氟乙酸盐的晶型b,在其x-射线粉末衍射(xrpd)图谱中,具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、14.0°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、22.2°±0.2°、23.5°±0.2°的衍射峰,典型地具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、12.4°±0.2°、14.0°±0.2°、14.9°±0.2°、16.1°±0.2°、16.6°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、21.1°±0.2°、21.8°±0.2°、22.2°±0.2°、23.5°±0.2°的衍射峰,更典型地具有2θ=7.0°±0.2°、10.2°±0.2°、11.7°±0.2°、12.4°±0.2°、13.3°±0.2°、14.0°±0.2°、14.9°±0.2°、16.1°±0.2°、16.6°±0.2°、17.3°±0.2°、18.6°±0.2°、19.1°±0.2°、20.2°±0.2°、21.1°±0.2°、21.8°±0.2°、22.2°±0.2°、23.1°±0.2°、23.5°±0.2°、24.4°±0.2°、24.7°±0.2°、26.1°±0.2°、28.2°±0.2°的衍射峰。本发明中,x-射线衍射光谱采用下述方法测定:仪器:brukerd8advancex-射线衍射仪;方法:靶:cu:k-alpha;波长管压voltage:40kv;管流current:40ma;扫描范围:4~40°;样品旋转速度:15rpm;扫描速度:10°/分钟。在本发明的部分实施方式中,本发明的式i所示化合物的三氟乙酸盐的晶型b的衍射峰具有如下特征:序号2θ±0.2(°)相对强度(%)序号2θ±0.2(°)相对强度(%)17.0100.01519.66.8210.242.41620.239.4311.732.01721.116.6412.421.31821.823.9513.37.11922.231.0614.040.12023.19.6714.916.22123.537.6816.118.12224.413.0916.619.92324.711.51016.98.32426.19.51117.310.22526.45.81217.66.22628.27.31318.634.42732.34.11419.141.7在本发明的部分实施方式中,所述式i所示化合物的三氟乙酸盐的晶型b的x-射线粉末衍射图谱基本上如图1所示。需要说明的是,在x-射线衍射光谱中,由结晶化合物得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。此外,对任何给定的晶型而言,峰的位置可能存在轻微误差,这在晶体学领域中也是公知的。例如,由于分析样品时温度的变化、样品移动、或仪器的标定等,峰的位置可以移动,2θ值的测定误差有时约为±0.2°,典型地约为±0.1°。因此,在确定每种晶型结构时,应该将此误差考虑在内。在xrd图谱中通常用2θ角或晶面距d表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d代表晶面距,λ表入射x射线的波长,θ为衍射角。对于同种化合物的同种晶型,其xrd谱的峰位置在整体上具有相似性,相对强度误差可能较大。还应指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线的缺失,此时,无需依赖高纯试样中观察到的全部谱带,甚至一条谱带也可能对给定的晶体是特征性的。本发明的式i所示化合物的三氟乙酸盐的晶型b也可以用dsc进行表征,起始温度为171.4℃±5℃,峰值温度为173.4℃±5℃。在本发明的部分实施方式中,所述式i所示化合物的三氟乙酸盐的晶型b的dsc谱图如图2所示。本发明中,差示扫描量热分析(dsc)采用下述方法测定:仪器:taq2000差示扫描量热仪;方法:取样品(~1mg)置于dsc铝锅内进行测试,方法为:25℃~300℃,升温速率10℃/min。dsc测定当晶体由于其晶体结构发生变化或晶体熔融而吸收或释放热时的转变温度。对于同种化合物的同种晶型,在连续的分析中,热转变温度和熔点误差典型的在约5℃之内,通常在约3℃之内,当我们说一个化合物具有某一给定的dsc峰或熔点时,这是指该dsc峰或熔点±5℃。dsc提供了一种辨别不同晶型的辅助方法。不同的晶体形态可根据其不同的转变温度特征而加以识别。需要指出的是对于混合物而言,其dsc峰或熔点可能会在更大的范围内变动。此外,由于在物质熔化的过程中伴有分解,因此熔化温度与升温速率相关。在本发明的部分实施方式中,所述式i所示化合物的三氟乙酸盐晶型b的式i所示化合物与三氟乙酸的摩尔比为1:0.5-2,优选1:1。本发明另一方面提供了三氟乙酸盐的晶型b的制备方法,包括使式i所示化合物的三氟乙酸盐从溶剂中析出。在本发明的部分实施方式中,该方法包括以下步骤:1)使式i所示化合物的三氟乙酸盐溶于结晶溶剂中,加热溶解;2)冷却析晶、洗涤、干燥。上述步骤1)中,所述结晶溶剂选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、丙酮、乙酸乙酯或它们混合溶剂、或者水;优选乙醇。上述步骤1)中,每1g式i所示化合物的三氟乙酸盐对应加入的结晶溶剂量为1~5ml,优选2~3ml。上述步骤1)中,所述加热溶解的目的是将式i所示化合物的三氟乙酸盐与结晶溶剂形成均相体系。本发明另一方面提供了包含上述晶型b的结晶组合物。在本发明的部分实施方式中,所述晶型b占结晶组合物重量的50%以上,优选80%以上,更优选90%以上,最优选95%以上。此外,所述“结晶组合物”中除了晶型b外还可以含有其它晶型、或无定形、或式i化合物以外的杂质。本发明的另一方面提供了一种药物组合物,该药物组合物中包含有效量的式i所示化合物的三氟乙酸盐、或所述晶型b、或者包含所述晶型b的结晶组合物,此外,该药物组合物还可以含有或不含有药学上可接受的载体、赋形剂和/或介质。本发明中,术语“药物组合物”是指本发明的晶型b或结晶组合物与在本领域中通常接受的用于将生物活性化合物输送至有机体(例如人)的载体、赋形剂和/或介质的制剂。药物组合物的目的是有利于对有机体给予本发明的式i所示化合物的三氟乙酸盐、其晶型b或结晶组合物。本发明中,术语“药学上可接受的载体”是指对有机体无明显刺激作用,而且不会损害该活性化合物的生物活性及性能的载体和稀释剂。“药学上可接受的赋形剂和/或介质”是指与活性成分一同给药的、有利于活性成分的给药的惰性物质。“药学上可接受的载体、赋形剂和/或介质”包括但不限于可用于人或家畜动物的任何载体、赋形剂、介质、助流剂、增甜剂、稀释剂、防腐剂、染料/着色剂、矫味增强剂、表面活性剂、润湿剂、分散剂、崩解剂、助悬剂、稳定剂、等渗剂、溶剂或乳化剂。所述赋形剂的非限制性实例包括碳酸钙、磷酸钙、各种糖和各类淀粉、纤维素衍生物、明胶、植物油和聚乙二醇等等。以纯的形式或以适宜的药物组合物形式的本发明化合物的给药可通过提供类似用途的药剂的任何可接受给药模式来进行。本发明的药物组合物可通过将本发明的化合物与适宜的药学上可接受的载剂、稀释剂或赋形剂组合而制备,且可配制成固态、半固态、液态或气态制剂,如片剂、丸剂、胶囊剂、粉剂、颗粒剂、膏剂、乳剂、悬浮剂、溶液剂、栓剂、注射剂、吸入剂、凝胶剂、微球及气溶胶等等。给予本发明化合物或其药物组合物的典型途径包括但不限于口服、直肠、透黏膜、经肠给药,或者局部、经皮、吸入、肠胃外、舌下、阴道内、鼻内、眼内、腹膜内、肌内、皮下、静脉内给药。本发明的药物组合物可以采用本领域众所周知的方法制造,如常规的混合法、溶解法、制粒法、制糖衣药丸法、磨细法、乳化法、冷冻干燥法等等。在优选的实施方式中,药物组合物是口服形式。对于口服给药,可以通过将活性化合物与本领域熟知的药物可接受的载体、赋形剂和/或介质混合,来配制该药物组合物。这些载体、赋形剂和介质能使本发明的化合物被配制成片剂、丸剂、锭剂、糖衣剂、胶囊剂、液体、凝胶剂、浆剂、悬浮剂等,用于对患者的口服给药。可以通过常规的混合、填充或压片方法来制备固体口服组合物。例如,可通过下述方法获得:将所述的活性化合物与固体赋形剂混合,任选地碾磨所得的混合物,如果需要则加入其它合适的辅剂,然后将该混合物加工成颗粒,得到了片剂或糖衣剂的核心。适合的赋形剂包括但不限于:填充剂,如糖,包括乳糖、蔗糖、甘露醇或山梨糖醇;纤维素制剂例如微晶纤维素、玉米淀粉、小麦淀粉、大米淀粉和马铃薯淀粉;以及其它物质,如硅胶、明胶、黄芪胶、甲基纤维素、羟丙基甲基纤维素、羟甲基纤维素钠和/或聚乙烯吡咯烷酮;崩解剂,如羧甲淀粉钠、交联羧甲基纤维素钠、交联聚乙烯吡咯烷酮、琼脂或藻酸,也可以使用盐,如藻酸钠。可以根据通常药物实践中公知的方法任选地对糖衣剂的核心进行包衣,尤其使用肠溶包衣。本发明的另一方面提供了式i所示化合物的三氟乙酸盐、晶型b或其结晶组合物在制备治疗toll样受体7(tlr7)相关疾病的药物中的用途,优选的,所述疾病选自病毒感染。本发明提供的式i所示化合物的三氟乙酸盐的晶型b具有纯度高、结晶度高、稳定性好等优点;可供制备用于预防或治疗toll样受体7(tlr7)相关疾病的药物。实施例本发明所使用的所有溶剂是市售的,无需进一步纯化即可使用。反应一般是在惰性氮气下、无水溶剂中进行的。质子核磁共振数据记录在brukeravanceiii400(400mhz)分光仪上,化学位移以四甲基硅烷低场处的(ppm)表示。质谱是在安捷伦1200系列加6110(&1956a)上测定。lc/ms或shimadzums包含一个dad:spd-m20a(lc)和shimadzumicromass2020检测器。质谱仪配备有一个正或负模式下操作的电喷雾离子源(esi)。实施例1:2-丁氧基-7-(4-(吡咯烷-1-基甲基)苄基)-5h-吡咯并[3,2-d]嘧啶-4-胺三氟乙酸盐的制备式iii:2,4-二氯-5-((2-(三甲基硅基)乙氧基)甲基)-5h-吡咯并[3,2-d]嘧啶式ii化合物(2,4-二氯-5h-吡咯并[3,2-d]嘧啶)(4.00kg,21.28mol)溶解于dmf(20.00l)中;室温(25℃)下分批加入dipea(2.58kg,20.00mol),随后搅拌30min,反应液用冰浴降温至0℃,然后在5小时内,以1~2滴/秒的滴速,缓慢滴加sem-cl(4.00kg,24.00mol),滴完后,反应液在0℃下搅拌反应4小时,hplc监测反应完全,反应液用70l水淬灭稀释后,用乙酸乙酯(15l×3)萃取,合并的有机相依次用1m的盐酸水溶液(5l×2)和饱和食盐水(7l×2)洗涤,减压蒸馏除去溶剂后得式iii化合物(6.40kg,20.11mol,产率94.50%)。1hnmr(400mhz,dmso-d6)δ8.24-8.35(m,1h),6.70-6.85(m,1h),5.77(s,2h),3.45-3.57(m,2h),0.74-0.86(m,2h),0.00(s,9h)。式iv:2-氯-5-((2-(三甲基硅基)乙氧基)甲基)-5h-吡咯并[3,2-d]嘧啶-4-胺10l高压釜中,式iii化合物(1.60kg,5.03mol)溶于异丙醇(1.60l),室温(25℃)下一次性加入氨水(4l),反应混合物在95℃下搅拌7小时,hplc监测反应完毕,反应液自然冷却到室温,经布氏漏斗过滤后得到黑褐色固体,该固体依次用乙酸乙酯/正庚烷(1/1,5l×2)中打浆,乙酸乙酯(4l)打浆,得到式iv化合物棕色固体(1.25kg,4.18mol,产率83.1%)。1hnmr(400mhz,dmso-d6)δ7.61-7.77(m,1h),6.97-7.19(m,2h),6.28-6.38(m,1h),5.54-5.67(m,2h),3.43-3.53(m,2h),0.76-0.91(m,2h),0.07(s,9h)。式v:2-丁氧基-5-((2-(三甲基硅)基乙氧基)甲基)-5h-吡咯并[3,2-d]嘧啶-4-胺氮气保护下,金属钠(525.05g,22.84mol)缓慢分批加入到n-buoh(17.0l)中;加入完毕后,体系升温至60℃,并在该温度下持续搅拌,直至金属钠全部溶解,随后,体系冷却至25℃,将式iv化合物(1.95kg,6.53mol)分批加入,搅拌混合均匀后,反应物在90℃下持续搅拌8小时,hplc监测反应完全,反应混合物自然降温至25℃后,缓慢倒入30l饱和氯化铵水溶液中,随后用乙酸乙酯(15l×3)萃取,合并的有机相用饱和食盐水(20l×2)洗涤,经无水na2so4干燥、过滤后,减压蒸馏除去溶剂后,残余物在正庚烷(4l)中打浆,过滤分离得到固体,再在乙酸乙酯(5l)中打浆,得到式v化合物黄白色固体(1.53kg,4.55mol,69.7%)。1hnmr(400mhz,dmso-d6)δ7.49-7.54(m,1h),6.54-6.62(m,2h),6.15-6.20(m,1h),5.54(s,2h),4.10-4.22(m,2h),3.42-3.55(m,2h),1.58-1.73(m,2h),1.35-1.47(m,2h),0.90-0.96(m,3h),0.83-0.89(m,2h),0.05(s,9h)。式vi:2-丁氧基-5h-吡咯并[3,2-d]嘧啶-4-胺式v化合物(1.10kg,3.27mol)溶于tfa(5.50l),反应液在25℃下持续搅拌16小时,hplc监测反应完全,减压蒸馏除去tfa,剩余物溶解在甲醇(1.2l)和冰水(1.2l)中,均匀搅拌下,用浓氨水调节体系ph至12,然后搅拌2小时,溶液中不断有沉淀析出,过滤后,滤饼为白色固体,依次用15%的氨水(1.2l×3)和乙酸乙酯(4l)打浆,得到式vi化合物白色固体(550.00g,2.67mol,81.7%)。1hnmr(400mhz,甲醇-d4)δ7.37(d,j=2.89hz,1h),6.29(d,j=3.01hz,1h),4.27(t,j=6.53hz,2h),1.75(d,j=7.91hz,2h),1.44-1.61(m,2h),1.00(t,j=7.40hz,3h)。式vii:4-((4-氨基-2-丁氧基-5h-吡咯并[3,2-d]嘧啶-7-基)-羟基甲基)苯甲醛将对苯二甲醛(790.64mg,5.82mmol)和异丙醇(10ml)投入至三口瓶中,搅拌下加入2-丁氧基-5h-吡咯并[3,2-d]嘧啶-4-胺(1.00g,4.85mmol),降温至0℃,之后继续搅拌10min,加入纯化水(10ml)和碳酸钾(804.17mg,5.82mmol),25℃反应16hr,lcms监控至原料反应完全,反应完全后,有固体析出。过滤,所得固体用20ml纯化水打浆,30ml(乙酸乙酯/正庚烷=1/20)打浆,过滤,干燥,得到式vii化合物黄色固体(1.50g,4.41mmol,收率:90.9%)。1hnmr(400mhz,甲醇-d4)δ9.94(s,1h),7.86(d,j=8.16hz,2h),7.72(d,j=8.16hz,2h),7.12-7.17(m,1h),6.19(s,1h),4.28(t,j=6.53hz,2h),1.68-1.77(m,2h),1.44-1.54(m,2h),0.97(t,j=7.34hz,3h)。式viii:(4-氨基-2-丁氧基-5h–吡咯并[3,2-d]嘧啶-7-基)(4-(吡咯烷-1-基甲基)苯基)甲醇将式vii化合物(450.0g,1.32mol)和异丙醇(4.5l)投入至30l反应釜中,搅拌5min,之后加入冰醋酸(119.0g,1.98mol),搅拌降温至0-10℃,滴加吡咯烷(112.4g,1.58mol),在滴加中保持温度低于10℃,滴毕,分批次加入三乙酰氧基硼氢化钠(420.0g,1.98mol),10-20℃反应3hr,液相监控至原料完全消失,反应完全后,加入纯化水5l,将溶液温度降至-10℃左右,加入15%氨水12l,加入过程中溶液温度低于0℃,搅拌有固体析出,过滤,所得滤饼2l水打浆,2l×2乙酸乙酯打浆,过滤,40℃减压烘干12hr,得到式viii化合物,黄色固体465.0g,1.18mol,收率89.4%,水分0.9%。1hnmr(400mhz,甲醇-d4)δ7.46(d,j=7.91hz,1h),7.29(d,j=8.03hz,1h),7.09(s,1h),6.12(s,1h),4.29(t,j=6.53hz,2h),3.60(s,2h),2.52(br.s.,4h),1.66-1.83(m,6h),1.49(d,j=7.53hz,2h),0.98(t,j=7.40hz,3h)。式i:2-丁氧基-7-(4-(吡咯烷-1-基甲基)苄基)-5h-吡咯并[3,2-d]嘧啶-4-胺将式viii化合物(440.0g,1.11mol)和二氯甲烷(7.0l)投入20l反应釜中,搅拌降温至-15℃以下,滴加三乙基硅烷(880ml,5.55mol)后,继续滴加三氟乙酸(880ml),在滴加中保持温度低于-10℃,滴加完毕后,0℃反应2hr,液相监控至原料点消失,反应完全后,将反应液浓缩至干,加入乙酸乙酯2.2l,搅拌降温至0℃以下,之后加入饱和碳酸钠溶液调节溶液ph=9-10,此过程中温度小于10℃,过滤,所得滤饼用2.2l水打浆后过滤,减压烘干,得到式i的三氟乙酸盐白色固体550g,其中式i所示化合物与三氟乙酸的摩尔比为1:1。1hnmr(400mhz,甲醇-d4)δ7.46(d,j=7.91hz,1h),7.29(d,j=8.03hz,1h),7.09(s,1h),6.12(s,1h),4.29(t,j=6.53hz,2h),3.60(s,2h),2.52(br.s.,4h),1.66-1.83(m,6h),1.49(d,j=7.53hz,2h),0.98(t,j=7.40hz,3h)。实施例2:2-丁氧基-7-(4-(吡咯烷-1-基甲基)苄基)-5h-吡咯并[3,2-d]嘧啶-4-胺三氟乙酸盐晶型b的制备。将实施例1所得白色固体和1.4l乙醇搅拌加热至回流,搅拌至全部溶解,过滤,滤液室温搅拌析晶,得到白色固体,之后重复上述操作,重结晶一次,得到类白色固体210.0g,即为式i所示化合物三氟乙酸盐晶型b,其中,式i所示化合物与三氟乙酸的摩尔比为1:1。采用下述方法对xrpd进行测定:仪器:brukerd8advancex-射线衍射仪;方法:靶:cu:k-alpha;波长管压voltage:40kv;管流current:40ma;扫描范围:4~40°;样品旋转速度:15rpm;扫描速度:10°/分钟。结果显示,所获得的化合物晶体具有基本上如图1所示的衍射峰。药物活性效果实施例效果实施例1:toll样受体7和toll样受体8体外受体结合活性筛选方案试剂:hek-bluehtlr7细胞和hek-bluehtlr8细胞(来源于invivogen公司)dmem培养基热灭活胎牛血清抗支原体试剂normocintm博来霉素杀稻瘟菌素方案:(1)96孔化合物板的准备:利用液体工作站pod将化合物从10mmol/l浓度起始,用dmso做3倍梯度稀释,共稀释10个点(从第2列到第11列,每个点2个重复)。在第12列加入1μl5mg/ml的阳性化合物r848作为阳性对照,在第1列加入1μldmso作为阴性对照。每孔中含有的dmso体积均为1μl。(2)收集细胞培养瓶中的细胞,将细胞密度稀释成250,000个细胞/ml。(3)加入200μl(50,000个细胞/孔)细胞悬液至准备好的化合物板中。每孔中dmso终浓度为0.5%。(4)将含有细胞和化合物的培养板放入co2培养箱中培养24小时,培养条件为37℃,5%co2浓度。(5)培养24小时后,从细胞培养板中每孔取出20μl上清液转移到一块96孔透明检测板中。然后往检测板中每孔加入180μlquanti-blue试剂,并置于37℃,5%co2培养箱孵育1小时。(6)1小时后,用酶标仪od650读板检测20μl上清液中碱性磷酸酶的含量。(7)利用prism软件分析数据,得出各化合物的ec50。实验结果如表1所示:表1化合物名称tlr7ec50(nm)tlr8ec50(nm)gs-96205177867式i化合物16011632结论:本发明的式i所示化合物展现出比对照品(toll样受体7激动剂gs-9620)更高的与toll样受体7体外受体结合活性,以及比对照品(toll样受体7激动剂gs-9620)更低的与toll样受体8体外受体结合活性。效果实施例2外周血单个核细胞试验方案本实验目的在于检测利用式i所示化合物刺激人外周血单核细胞(pbmc)24h后细胞因子的表达水平。检测时细胞上清液不稀释,直接检测ifn-α的水平。实验过程中首先将式i的化合物配制成20mmol浓度的dmso储存液,用细胞培养基做10倍梯度稀释,总共稀释11个点。取其中9个稀释点的化合物(化合物的最高浓度为200μmol/l)加入96孔板中,每孔50μl,然后接种新鲜的人外周血单核细胞,每孔接种150μl体系,其中含有450,000个细胞。将细胞培养板置于37℃和5%co2培养箱中培养24小时,培养结束后将培养板以1200rpm的速度离心5分钟,随后收集上清并将其储存于-20℃以待检测。细胞因子的检测利用bd公司的流式液相多重蛋白定量技术(cba),在流式细胞仪上完成检测。利用上述检测方法,我们将刺激产生30pg/ml的ifn-α的最低药物浓度,定义为该化合物在该细胞因子刺激实验上的mec值。实验结果如下表2所示:表2化合物名称inf-αmec(nm)tnf-αmec(nm)gs-962050500式i的化合物5500结论:本发明的式i化合物展示出比对照品(gs-9620)更好的体外pbmc的ifn-α诱导活性、以及与对照品(gs-9620)相当的tnf-α的诱导活性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1