利用优化的基因密码子扩展系统通读提前终止密码子疾病中的截短蛋白的制作方法
本发明属于生物制药领域,具体涉及利用基因密码扩展的非天然氨基酸系统,通读单基因遗传病的无义突变位点。并且通过改造古甲烷球菌的trna(trnapyl),得到全新的高通读效率的uaa和uga编码的非天然氨基酸系统,扩展了trnapyl和吡咯赖氨酰-trna合成酶(pylrs)正交对的使用范围,可用于通读三种无义突变的终止密码子uag,uaa和uga。
背景技术:
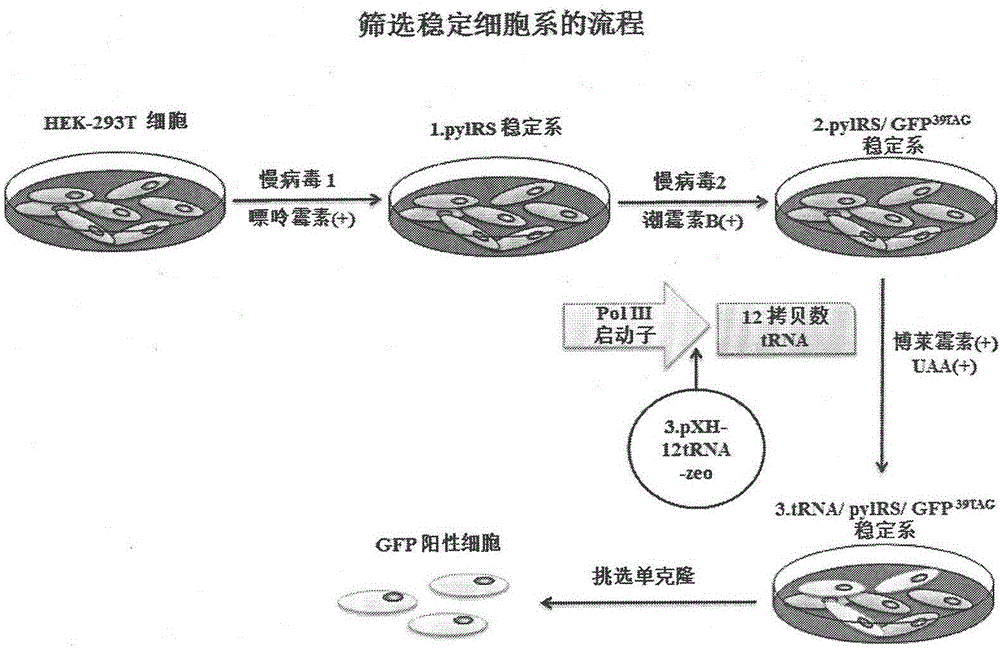
:无义突变导致的遗传性疾病人类基因组中的存在许多种基因突变类型,无义突变是基因突变中的一类。基因突变是基因组dna分子发生的可遗传的变异现象,其中包括移码突变和碱基置换。移码突变包括碱基的插入和缺失,碱基置换主要为错义突变和无义突变。无义突变是指编码基因的某个碱基发生突变,产生终止密码子uag、uaa和uga,终止密码子不编码任何氨基酸。终止密码子不能与转移rna(trna)的反密码子配对,但能被终止因子或释放因子识别,终止肽键的合成,使蛋白质合成终止,因而生成不完整和没有功能的蛋白质。无义突变的发生使基因框内产生提前终止密码子(prematureterminationcodons,ptc),导致基因编码的两种结果,一种即产生截短型蛋白,另一种则导致含有ptc的mrna的稳定性降低,从而引发无义介导的mrna降解途径(nmd)。据统计,大约有11.2%的遗传性疾病会产生ptc突变,被称作提前终止密码子病(prematureterminationcodonsdiseases,ptcdiseases),另一方面,许多癌症发生也会产生ptc突变(keelingk.m.,wangd.,conards.e.,bedwelld.m..suppressionofprematureterminationcodonsasatherapeuticapproach.criticalreviewsinbiochemistryandmolecularbiology,2012,47:444-463.)。杜氏肌营养不良(duchennemusculardystrophy,dmd)是ptc疾病中 的一个典型代表。dmd是一种严重的肌肉萎缩疾病,也是最常见的x连锁隐性遗传性疾病,以进展性、致死性为主要特点。dmd基因的无义突变则是导致dmd发生的主要原因之一。无义突变产生提前终止密码子uag、uaa、uga,生成截短了的多肽产物,使患者缺失或缺少功能性的抗萎缩蛋白(dystrophin),致使肌肉萎缩。据报道,duchenne型肌营养不良症在活产男婴中的发病率为1/6300~1/3500[dooleyj,gordonke,doddsl,macsweenj.duchennemusculardystrophy:a30-yearpopulation-basedincidencestudy.clinpediatr(phila),2010,49:177-179.]。该病目前尚无有效治愈方法,多于幼年期发病,青少年期丧失行走能力,成年早期死亡,给患者个人、家庭和社会造成沉重心理和经济负担。先前的研究中通读提前终止密码子的方法有:(1)化学小分子诱导的通读:氨基糖苷类药物如g418和非氨基糖苷类药物如ptc124。1996年,howard等在对囊性纤维变性的研究过程中首次观察到氨基糖苷类抗生素可以在哺乳动物细胞内诱导ptc通读,从而合成完整的有功能的蛋白质。但氨基糖苷类抗生素在发挥无义抑制作用的同时,可产生严重的不良反应,其中最严重的是耳毒性和肾毒性。并且在2016年2月,ptc124刚被美国fda拒绝受理(2)外显子跳跃方法:针对跳过外显子51表达dmd患者的反义核苷酸药物。但fda已经拒绝了biomarin的drisapersen。而另一家公司sareptatherapeutics的eteplirsen在2016年5月将会得到fda的评审结果;(3)抑制子trna通读:其反密码子环发生突变而能和终止密码子配对,以致能通读终止密码子。该治疗手段难以进入临床应用的主要原因suppressortrna可能会识别正常终止密码子从而导致异常蛋白的潜在毒性。遗传密码扩展技术经过数年的研究,人们对原核生物核糖体的翻译机制已有较全面的理解,多种核糖体不同功能状态的晶体和电镜结构已得到解析,大多数氨trna合成酶的结构也已获得。基于这些研究成果,近年来发展起来了遗传密码扩展的技术-利用琥珀终止密码子(tag)来编码多种非天然氨基酸并在生物活体内将其定点插入。到目前为止,这一技术已经将数种非天然氨基酸成功地定点表达在活细胞的蛋白质当中,赋予了这些蛋白质新颖的物理、化学和 生理性质。使用这一方法,可以将非天然氨基酸(包括亲和标记和光致异构化的氨基酸、羰基氨基酸和糖基化氨基酸)引入蛋白质中(l.wang等人,(2001),science292:498-500;j.w.chin等,2002,journaloftheamericanchemicalsociety124:9026-9027;j.w.chin,&p.g.schultz,2002,chembiochem11:1135-1137)。这些研究表明,有可能且有选择性且常规地引入化学官能基团到蛋白质中,例如,羰基、炔基、和叠氮基团等特殊化学基团,这些基团一般能够有效且选择性地形成稳定的共价键,引入致病蛋白后可以用于研究致病蛋白和其他蛋白相互作用的机制。经观察trnapyl和pylrs合成酶的复合体的晶体结构,pylrs合成酶不识别trnapyl的反密码子环,所以我们认为改变trnapyl的反密码子环的碱基序列不影响trnapyl和pylrs合成酶的正交性。技术实现要素:发明人经过对现有技术的思考和研究,通过改造古甲烷球菌的trna(trnapyl),构建了pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒,得到全新的高通读效率的uaa和uga编码的非天然氨基酸系统,使之能够用于通读三种终止密码子uag,uaa和uga;构建了模拟内源性提前终止密码子的质粒——在由内含子和外显子组成的smad基因上引入提前终止密码子,可以用于评价通读内源性提前终止密码子的效率;将基因密码子扩展技术用于通读单基因遗传病的和肿瘤细胞中抑癌基因的无义突变位点,恢复相应蛋白质的表达。本发明的优点可体现在如下中的一个或几个:1、获得了全新的高通读效率的uaa和uga编码的非天然氨基酸系统。2、利用基因密码子扩展技术,实现通读遗传性疾病中的无义突变,恢复截短蛋白的正常结构和功能。具体地,在本发明的一个具体的实施方案中,构建了识别三种终止密码子(amber,ocher,opal)的三种trnapyl/pylrs质粒,并在稳定细胞系hek293-pyl(已保藏于中国普通微生物菌种保藏管理中心,保藏日为2015年11月17日、保藏号为cgmccno:11592。其分类命名为人hek293t细胞)中 恢复dmd疾病蛋白dystrophin的表达、通读了内源性提前终止密码子和在a549和du145肿瘤细胞系中恢复了抑癌基因stk11和ephb2蛋白的表达。主要通过以下6个步骤:(1)构建pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒;(2)构建含有提前终止密码子的gfp报告基因pcdna3.1-gfp-39tag;pcdna3.1-gfp-39taa;pcdna3.1-gfp-39tga;(3)根据dmd病人的无义突变位点,利用点突变技术在dystrophin蛋白的同源异构体蛋白dp71b蛋白相应位点引入提前终止密码子,构建含有提前终止密码子uag的dp71b蛋白质粒dp71b3116tag,dp71b3317tag,dp71b3601tag;(4)在smad基因(由内含子和外显子组成)上引入提前终止密码子tag,构建了模拟内源性提前终止密码子的质粒pcdna3-smad-39tag,pcdna3-smad-122tag和pcdna3-smad-133tag;(5)将步骤(1)和(2)的质粒交叉对应转染至293t细胞,加入非天然氨基酸培养48小时后观察绿色荧光,比较三种终止密码子的通读效率;(6)将步骤(3)中的质粒转染稳定细胞系hek293-pyl,加入非天然氨基酸培养48小时后提蛋白,westernblot方法检测到dp71b全长蛋白,疾病蛋白表达恢复;(7)将步骤(4)中的质粒转染稳定细胞系hek293-pyl,加入非天然氨基酸培养48小时后提蛋白,westernblot方法检测smad全长蛋白,证明基因密码子扩展技术能够有效抑制无义介导的mrna降解途径,通读不同位置内源性的提前终止密码子;(8)将pcmv-cua(trnapylcua/pylrs)转染肿瘤细胞系a549和du145,加入非天然氨基酸培养48小时后提取蛋白,经westernblot证明在肿瘤细胞系a549和du145中stk11蛋白和全长ephb2蛋白恢复表达。在本发明的一个具体的实施方案中,以pcmv-cua(trnapylcua/pylrs)作为模板质粒,设计点突变引物,利用定点突变试剂盒,将trnapylcua反密码子环上的碱基cua通过上述引物进行点突变为uua和uca,获得pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒。在本发明的一个具体的实施方案中,利用含有提前终止密码子的gfp绿色荧光蛋白检测三种trnapylcua/uua/uca/pylrs的通读效率。第一步利用点突变技术将gfp荧光基因第39位氨基酸密码子分别点突变为uag、uaa、uga 三种提前终止密码子得到pcdna3.1-gfp-39tag、pcdna3.1-gfp-39taa和pcdna3.1-gfp-39tga三个质粒。第二步用pcmv-cua/uua/cua和pcdna3.1-gfp-39tag/taaa/tga交叉对应共转染293t细胞。第三步加入非天然氨基酸培养48小时后用荧光显微镜观察绿色荧光。最终确认trnapyl/pylrs对与之完美配对的终止密码子有高效的通读作用,其中通读效率uag最高,uga次之,uaa最低。在本发明的一个具体的实施方案中,将基因密码子扩展技术应用于恢复人类遗传性疾病相关无义突变蛋白表达。根据人类dmd疾病中无义突变位置,在野生型dp71b序列对应位置进行点突变,构建含有提前终止密码子uag的dp71b蛋白质粒dp71b3116tag(c.9346c>t),dp71b3317tag(c.9952c>t),dp71b3601tag(c.10801c>t)。将质粒转染稳定细胞系hek293-pyl,加入非天然氨基酸培养48小时后提蛋白,westernblot方法检测到dp71b全长蛋白,疾病蛋白表达恢复。在本发明的一个具体的实施方案中,用稳定细胞系hek293-pyl验证trnapylcua/pylrs通读不同位置的内源性提前终止密码子。第一步将由内含子和外显子组成的smad基因克隆到pcdna3质粒上,然后利用点突变方法将smad第39位,122位和133位氨基酸密码子突变为uag提前终止密码子,得到质粒pcdna3-smad-39tag,pcdna3-smad-122tag和pcdna3-smad-133tag。将内源性提前终止密码子质粒(pcdna3-smad-39tag,pcdna3-smad-122tag或pcdna3-smad-133tag)顺时转染含有古甲烷球菌的trna(trnapyl)和吡咯赖氨酰-trna合成酶(pylrs)的稳定细胞系。加入非天然氨基酸后培养48小时后提取蛋白,经westernblot三组都检测到全长smad的蛋白。证明基因密码子扩展技术能够有效抑制无义介导的mrna降解途径,通读不同位置内源性的提前终止密码子,恢复全长蛋白的表达。在本发明的一个具体的实施方案中,将基因密码子扩展技术用完通读肿瘤细胞中抑癌基因的无义突变位点。将pcmv-cua(trnapylcua/pylrs)转染肿瘤细胞系a549和du145(人肺癌细胞a549基因组上stk11发生无义突变c.109c>t,p.q37x,为终止密码子uag;人前列腺癌细胞du145基因组上ephb2基因发生无义突变c.2167c>t,p.q723x,为终止密码子uag)。 加入非天然氨基酸培养48小时后提取蛋白,经westernblot证明基因密码子扩展技术在肿瘤细胞系a549和du145中恢复了全长stk11蛋白和全长ephb2蛋白的表达。更为具体地,本发明提供了1.利用基因密码子扩展技术,在无义突变体蛋白的提前终止密码子处插入非天然氨基酸来,通读恢复单基因遗传病中的致病蛋白和肿瘤细胞内抑癌基因蛋白的正常表达和功能的方法。2.如项目1所述的基因密码子扩展技术,由源自古甲烷球菌的trna(trnapyl)、吡咯赖氨酰-trna合成酶(pylrs)和非天然氨基酸组成。所述非天然氨基酸选自:所示的lys-diazirine(naek),所示的lys-azido,或其它含有双吖丙啶、叠氮结构的非天然氨基酸中的至少1种。3.如项目2所述的古甲烷球菌的trna(trnapyl),由原始trnapylcua的反密码子环定点突变改造成trnapyluua和trnapyluca,序列分别对应seqidno:1和seqidno:2,其特征在于分别与终止密码子uaa、uga完美配对。trnapyluua和trnapyluca分别构建在pcmv-uua和pcmv-uca质粒上。4.如项目1所述的致病蛋白或抑癌基因蛋白,其中所插入的非天然氨基酸是位于第n位的lys-diazirine,其在蛋白中的连接方式如下式所示:5.其中,由r1到r2的方向为氨基酸序列的n末端到c末端方向,第n位可 以是致病蛋白或抑癌基因蛋白上的任意一位氨基酸,相应地,r1为第1至第n-1位氨基酸残基,r2为第n+1位至c末端的氨基酸残基,r3为6.如权利项目1所述的致病蛋白或抑癌基因蛋白,其中所引入的非天然氨基酸是位于第n位的lys-azido,其在流感病毒蛋白中的连接方式如下式所示:其中,由r1到r2的方向为氨基酸序列的n末端到c末端方向,第n位可以是如权利要求1所述的致病蛋白或抑癌基因蛋白上的任意一位,相应地,r1为第1至第n-1位氨基酸残基,r2为第n+1位至c末端的氨基酸残基,r4为7.如项目1-5所述的基因密码子扩展技术,其通读效率是用内源性提终止密码子质粒pcdna3-smad表达smad蛋白的量评价。通过以下步骤:(1)将原序列如seqidno:3所示的smad基因克隆到pcdna3上。(2)将39,122,133位密码子突变为uag琥珀型终止密码子,突变体质粒pcdna3-smad-39tag,pcdna3-smad-122tag和pcdna3-smad-133tag。序列如seqidno:4~6所示。(3)突变体质粒转染稳定细胞系hek293-pyl,加入非天然氨基酸培养48小时后提取蛋白,经westernblot检测到全长smad蛋白。8.权利要求7所述的可稳定表达trna(trnapyl)和吡咯赖氨酰-trna合成酶(trnapyl)的哺乳动物稳定细胞系,其为hek293-pyl,保藏日为 2015年11月17日、其保藏号为cgmccno:11592。附图说明:图1.trnapylcua/pylrs,trnapyluua/pylrs和trnapyluca/pylrs分别通读gfp绿色荧光蛋白tag,taa和tga终止密码子。图2a.正交trna/氨酰trna合成酶的稳定细胞系hek293-pyl筛选方法的建立。图2b.双病毒过表达体系的构建。图2c.pxh-12t-zeo载体的构建。图3.westernblot验证在稳定细胞系hek293-pyl中,通读疾病蛋白dystrophin上的提前终止密码子从而恢复蛋白表达。图4.稳定细胞系hek293-pyl中通读内源性提前终止密码子图5.基因密码子扩展技术通读a549和du145肿瘤细胞系中提提前终止密码子,恢复stk11和ephb2蛋白表达为了更好地理解本发明,发明人用实施例对具体试验进行阐述和说明,其中所述实施例仅用于说明,并不限定本发明的保护范围。任何与本发明等价的变体或者实施方案都包括在本发明中。实施例1:pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒的构建(1)古甲烷球菌pcmv-cua质粒(trnapylcua/pylrs)的获得从保藏地:中国普通微生物菌种保藏管理中心菌种保藏地址:地址:北京市朝阳区北辰西路1号院,中国科学院微生物研究所,保藏日为2011年6月14日、保藏号为cgmccno:4951的分类命名为大肠埃希氏菌(escherichiacoli)的含有质粒pacyc-trna/pylrs的大肠埃希氏菌pacyc-trna/pylrs中获取质粒pacyc-trna/pylrs(以下简称该质粒为pcmv-cua),该质粒可以表达特异识别非天然氨基酸lys-diazirine和lys-azido的trna合成酶(pylrs)和特异性识别琥珀终止密码子uag的trna (trnapylcua)。(2)点突变trnapylcua构建pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒发明人针对突变trnapylcua的反密码子环,分别设计突变引物,具体的引物如下所示。表1trnapylcua的反密码子环点突变引物pcmv-uag-uaa-fortgtagatcgaatggactttaaatccgttcagccggpcmv-uag-uaa-revccggctgaacggatttaaagtccattcgatctacapcmv-uag-uga-forcatgtagatcgaatggacttcaaatccgttcagccgggttpcmv-uag-uga-revaacccggctgaacggatttgaagtccattcgatctacatg以pcmv-cua作为模板质粒,利用定点突变试剂盒(lightningsite-directedmutagenesiskits,catalog#210518),按说明书操作将trnapylcua反密码子环上的碱基cua通过上述引物进行点突变为uua和uca,获得pcmv-uua(trnapyluua/pylrs)和pcmv-uca(trnapyluca/pylrs)质粒,经测序验证突变成功。trnapyluua的序列为seqidno:1所示;trnapyluca的序列为seqidno:2所示。实施例2:利用含有提前终止密码子的gfp绿色荧光蛋白检测三种trnapylcua/uua/uca/pylrs正交系统的通读效率(1)非天然氨基酸lys-diazirine的合成和鉴定非天然氨基酸lys-diazirine的化学合成反应式如下。如上式所示,将原料1(5-羟基-2-戊酮)15ml与液氨40ml在-40℃下搅拌反应5h,之后降温至-60℃,缓慢滴加nh2oso3h的甲醇溶液,加毕升至室温,反应过夜。滤除沉淀,向上清液中加入三乙胺,冰浴条件下缓慢加入i2,至反应液颜色变深,不再产生气泡为止。反应完全后蒸除溶剂,经乙醚萃取后干燥。蒸除乙醚,剩余液体减压蒸馏获得25.4g无色粘稠液体产物2。将上述产物2用吡啶溶解,0℃搅拌下加入11gtscl,反应过夜。待反应完全后将反应液倒入浓盐酸与冰水的混合液中,乙醚萃取,醚层分别用1n盐酸和1nnaoh洗涤。有机相干燥柱分得到11.8g无色粘稠液体产物3。将上述产物3用dmf溶解,加入nan3室温反应隔夜至反应完全,加入大量水,乙醚萃取。蒸除乙醚,剩余产物用thf∶水(9∶1)混溶,加入三苯基磷,室温反应。反应完后加1nhcl混匀,旋干thf,二氯甲烷把未反应的原料,pph3和o=pph3洗掉,液相加1nnaoh调ph到12,二氯甲烷萃取出4.0g产物4。将5.2g原料5(boc-lys-ome)与羰基二咪唑反应,制备出5.9g化合物6。之后化合物6与上述产物4(4.0g)偶联得到化合物7,最后经过两步脱保护,将boc和甲酯脱除,得到目标4.5g产物8,即lys-diazirine。经谱学验证,结果为:1hnmr(400mhz,d2o):δ3.10(1h,t,j=6.3hz),2.96(4h,m),1.25(10h,m),0.90(3h,s);13cnmr(100mhz,d2o):183.63,160.66,56.00,39.80,39.30,34.49,30.84,29.20,26.75,23.92,22.43,18.80;hreimsm/z308.16937[m+1]+(calcdforc12h22n5nao3,308.16931),证明所得到的lys-diazirine结构正确。(2)构建含有提前终止密码子的gfp报告基因绿色荧光蛋白gfp是最常用的报告基因,也是指示非天然氨基酸插入的有力工具,其由238个氨基酸组成,其基因序列如seqidno:7。将gfp序列插入pcdna3.1商业质粒上,将gfp荧光基因第39位氨基酸密码子分别点突变为uag、uaa、uga三种提前终止密码子。设计能够使编码所述氨基酸的密码子分别突变为三种终止密码子的引物,具体引物如下表所示。表2gfp突变引物列表gfp-39-uag-forggcgagggcgatgccacctagggcaagctgaccctgaagttcgfp-39-uag-forgaacttcagggtcagcttgccctaggtggcatcgccctcgccgfp-39-uaa-forggcgagggcgatgccacctaaggcaagctgaccctgaagttcgfp-39-uaa-forgaacttcagggtcagcttgccttaggtggcatcgccctcgccgfp-39-uag-forggcgagggcgatgccacctgaggcaagctgaccctgaagttcgfp-39-uag-forgaacttcagggtcagcttgcctcaggtggcatcgccctcgcc利用定点突变试剂盒(lightningsite-directedmutagenesiskits,catalog#210518),按说明书操作,以野生型gfp表达载体pcdna3.1-gfp-wt为模板将第39位的氨基酸密码子分别突变为三种终止密码子,构建得到表达质粒(pcdna3.1-gfp-39tag、pcdna3.1-gfp-39taa和pcdna3.1-gfp-39tga),经测序验证突变成功。(3)293t细胞中顺转pcmv和pcdna3.1-gfp质粒验证突变后的正交系统的通读效率将实施例2的步骤2获得的pcdna3.1-gfp,以及实施例1的步骤2的pcmv质粒按表3的分组以1∶2比例混合,再与转染试剂 megatrans1.0按1∶3比例混合,共同加入293t细胞,6小时后换液加入浓度为1mm的naek,至细胞于37℃,5%co2的孵箱中继续培养48小时后后用荧光显微镜观察绿色荧光,结果如图1所示。最终确认trnapyl/pylrs对与之完美配对的终止密码子有高效的通读作用,其中通读效率uag最高,uga次之,uaa最低。表3pcmv质粒和gfp质粒分组混合组别质粒1pcmv-tag和pcdna3.1-gfp-39tag2pcmv-taa和pcdna3.1-gfp-39tag3pcmv-tga和pcdna3.1-gfp-39tag4pcmv-tag和pcdna3.1-gfp-39taa5pcmv-taa和pcdna3.1-gfp-39taa6pcmv-tga和pcdna3.1-gfp-39taa7pcmv-tag和pcdna3.1-gfp-39tga8pcmv-taa和pcdna3.1-gfp-39tga9pcmv-tga和pcdna3.1-gfp-39tga实例3:稳定细胞系hek293-pyl中通读疾病蛋白dystrophin(1)稳定细胞系hek293-pyl的构建构建了分别带有puromycin和hygromycin抗性的2个慢病毒过表达载体,两者分别携带氨酰trna合成酶和带有39位tag突变的报告基因gfp,通过两轮病毒转导hek-293t细胞和puromycin/hygromycin筛选,得到稳定细胞株pylrs/gfp39tag。之后,构建了携带12个拷贝数trna和zeomycin抗性的pxh-zeo-12trna载体,质粒线性化转染细胞株pylrs/gfp39tag,后经uaa存在下zeomycin筛选,最后分离出gfp阳性细胞(uaa存在下细胞呈绿色,去除uaa细胞呈无色),从而得到了表达正交trna/氨酰trna合成酶的稳定细胞系hek293-pyl(图2a)。a.载体的构建我们首先构建了分别带有puromycin和hygromycin抗性的2个慢病毒过表达载体,两者分别携带氨酰trna合成酶和带有39位tag突变的报告基因gfp,见图2b。我们从psd31载体出发,我们先通过bamhi/xbal酶切位点将sv40-puror基因分别替换成ires-puror和ires-hygror基因,这样就得到了2个不同抗性的病毒载体psd31-ires-puror和psd31-ires-hygror。其中,ires为内部核糖体进入序列(internalribosomeentrysite),ires序列常用于多顺反子基因表达。例如,在目的基因之后插入ires序列,后面是选择标记基因,这样转录出来的mrna就可以同时表达两种蛋白。利用ires系统过表达目的基因有2个优势:1.目的基因与标记基因共用一个启动子,避免了假阳性的出现;2.ires翻译效率低于传统翻译起始位点,使得目的基因表达量高于标记基因。所以,我们在ires位点的前面通过bamhi酶切位点分别引入cmv-pylrs序列和cmv-gfp39tag序列,就得到了能同时过表达两个目的蛋白的双病毒体系psd31-cmv-pylrs-ires-puror/psd31-cmv-gfp39tag-ires-hygror。所用主要引物见表4。表4.双病毒构建引物发明人用质粒稳定转染的方法过表达trna。为了保证trna的表达量,发明人构建得到了载体pxh-12t-zeo,其序列如seqidno:8所示。(图2c)b.慢病毒的包装和转导先包装psd31-cmv-pylrs-ires-puror病毒,转导hek293t细胞,puromycin筛选浓度为0.6ug/ml,得到稳定细胞系1号后,再加入psd31-cmv-gfp39tag-ires-hygror病毒,hygromycin筛选浓度为200ug/ml,得到稳定细胞系2号。c.质粒的稳定转染发明人通过质粒稳定转染,进行了第三轮筛选,最后得到了稳定表达正交trna/氨酰trna合成酶的特殊细胞系,步骤如下:a.将pxh-12t-zeo载体酶切线性化后,转染表达pylrs和gfp39tag蛋白的稳定细胞系2号(10cm培养皿,每皿10ug质粒,转染时不能有抗生素的存在)。b.转染6小时后换液,加入非天然氨基酸。c.转染48小时后,观察绿色荧光,换液,加入400ug/ml的zeomycin。d.每3天换液,直到blank组全部死亡,转染组形成克隆。e.分离纯化gfp阳性克隆,继续用剂量减半的zeomycin扩大培养,得到12t-zeo稳定细胞系hek293-pyl。质粒稳定转染筛选单克隆的要点如下:a.质粒稳定转染细胞密度很重要,筛选时细胞密度偏稀,易死亡难形成克隆。b.从单克隆化时开始,就要加大营养,血清和生长因子。c.单克隆接种到孔中细胞数目很少时,细胞之间的信号会变得很弱,也会导致阳性细胞的状态不佳甚至死亡。可以使用一种特殊的培养液:即细胞汇合度达到80%的时候的旧培养液通过滤器消毒,和新鲜的培养液按1∶1混合备用。或者适当增加血清浓度。d.单克隆消化后,不要加zeomycin和uaa,等细胞贴壁后再加,避免细胞死亡。(2)构建含有提前终止密码子uag的dp71b突变质粒dystrophin蛋白的同源异构体dp71b序列如seqidno:9所示,发明人根据杜氏肌营养不良病人无义突变的位点,对野生型dp71b序列进行点突变,在不同位置引入提前终止密码子,构建含有提前终止密码子uag的dp71b质粒dp71b3116tag(c.9346c>t),dp71b3317tag(c.9952c>t),dp71b3601tag(c.10801c>t),如序列seqidno:10~12所示,经测序验证突变成功。表5:dp71b点突变引物dp71b-9346-fortgaaactccgaagactgtagaaggccctttgcttgdp71b-9346-forcaagcaaagggccttctacagtcttcggagtttcadp71b-9952-forcatcaggccaaatgtaacatctgcaaatagtgtccaatcattdp71b-9952-foraatgattggacactatttgcagatgttacatttggcctgatgdp71b-10801-forgctggagcaaccctaggcagaggccaadp71b-10801-forttggcctctgcctagggttgctccagc(3)稳定细胞系hek293-pyl中通读疾病蛋白dystrophin将实施例3的步骤2获得的dp71b3116tag,dp71b3317tag,dp71b3601tag质粒与转染试剂megatrans1.0按1∶3比例混合,共同加入稳定细胞系hek293-pyl中,6小时后换液加入浓度为1mm的naek,至细胞于37℃,5%co2的孵箱中继续培养48小时后提取蛋白,westernblot检测(一抗为anti-dystrophin,为抗dystrophin蛋白c末端抗体,货号12715-1-ap)到全长dystrophin蛋白的产生,如图3。证明trnapyl/pylrs能够通读不同位置的提前终止密码子,恢复疾病蛋白的表达。实施例4:稳定细胞系hek293-pyl中通读内源性提前终止密码子效果考察(1)内源性提前终止密码子质粒pcdna3.1-smad-39tag;pcdna3.1-smad-39taa;pcdna3.1-smad-39tga构建将有内含子和外显子组成的smad基因序列(如seqid:3所示)插入 pcdna3.1商业质粒上,然后将smad第39位,122位和133位氨基酸密码子突变为uag提前终止密码子,得到质粒pcdna3-smad-39tag,pcdna3-smad-122tag和pcdna3-smad-133tag(如seqid:4~6所示)。(2)稳定细胞系中验证内源性提前终止密码子的通读将实施例4的步骤1获得的pcdna3-smad-39tag,pcdna3-smad-122tag或pcdna3-smad-133tag质粒与转染试剂megatrans1.0按1∶3比例混合,加入稳定细胞系hek293-pyl中,6小时后换液加入浓度为1mm的naek,至细胞于37℃,5%co2的孵箱中继续培养48小时后提取蛋白,westernblot检测(一抗为anti-myc,标签抗体)到全长smad蛋白的产生,如图4。验证基因密码子扩展技术能够抑制无义介导的mrna降解过程,并通读提前终止密码子,恢复蛋白的表达。实施例5:基因密码子扩展通读肿瘤细胞系基因组上的提前终止密码子经查阅文献,人肺癌细胞a549基因组上stk11发生无义突变c.109c>t,p.q37x,为琥珀型终止密码子uag;人前列腺癌细胞du145基因组上ephb2基因发生无义突变c.2167c>t,p.q723x,为琥珀型终止密码子uag。将pcmv-cua(trnapylcua/pylrs)质粒与转染试剂megatrans1.0按1∶3比例混合,分别转染a549和du145细胞,6小时后换液加入浓度为1mm的naek,至细胞于37℃,5%co2的孵箱中继续培养48小时后提取蛋白,westernblot检测(一抗分别为anti-stk11和anti-ephb2)到全长蛋白的stk11和ephb2产生,如图5。验证基因密码子扩展技术能够通读内源性基因组上的提前终止密码子,恢复抑癌基因蛋白表达。以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1