利用CRISPR技术编辑CCR5基因的sgRNA序列及其用途的制作方法
本发明涉及生物技术与基因治疗领域,尤其涉及利用crispr系统靶向敲除ccr5基因和治疗艾滋病的sgrna序列及其用途。
背景技术:
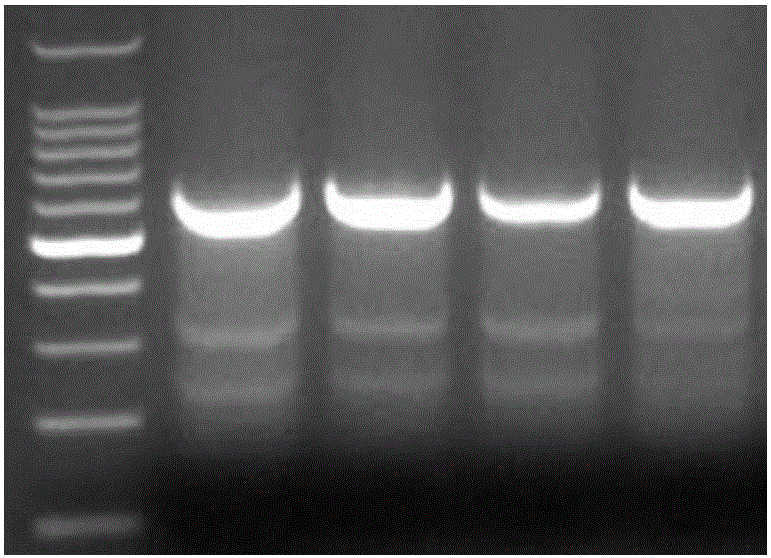
:crispr/cas系统是一种细菌和古细菌用于外源遗传物质的降解的适应性免疫防御机制。在这些生物体中,来自噬菌体的外源遗传物质获得和整合入crispr位点。这种新材料,也称为间隔区,建立用于将来对噬菌体感染性的序列特异性的片段。这些序列特异性的片段被翻译成引导crispr的rna(sgrnas),其功能为经由crispr相关(cas)蛋白的核酸酶活性,由crispr位点编码导向,互补侵入dna裂解。ii型crispr系统cas9核酸酶具有rna结合域,α螺旋识别叶(rec),核酸叶,其中包括ruvc和hnh的dna裂解和前间区序列邻近基序(pam)交叉位点。sgrna形成与cas9核酸酶的复合通过结合到rec叶内桥螺旋,并形成多个具有sgrna骨干的盐桥。一旦sgrna结合到cas9,cas9核酸酶的构象发生改变,产生一条能通道,让dna更容易结合。cas9/sgrna复合体扫描dna找到pam(5'-ngg)位点。识别pam位点导致dna解旋,使sgrna找到pam位点相邻的dna互补链。当cas9结合到pam位点相邻的,与sgrna互补的dna序列上,rec叶内部的桥螺旋和靶dna形成rna-dna异源双链结构。pam位点的识别包括可使靶dna双链断裂(dsb)的hnh和ruvc核断裂的激活,和导致dna降解。如果sgrna与靶dna不互补,cas9将会释放出来,寻找新的pam位点。dna中的线性靶基因组断裂后可以通过非同源末端接合(nhej)或者同源介导的修复(hdr)来进行修复。非同源末端接合(nhej)会引起插入或者删除错误。同源介导的修复(hdr)可在基因组的特异性位点结合特定标记。crispr/cas9机制可用于包括哺乳动物细胞在内的各种系统的基因工程中。如今,crispr/cas9系统在基因治疗领域大放异彩,新近报道的利用crispr/cas9系统对先天性肌营养不良的基因编辑为病人恢复正常的肌肉功能更是获得领域内的大量关注。在crispr/cas9应用于临床的前景被一致看好的当下,越来越多的crispr/cas9系统优化方案被提出。近年来,艾滋病治疗研究取得了突破性进展。2007年,德国一个医疗小组将ccr5基因缺陷的造血干细胞移植给一名患有白血病的艾滋病患者。通过四年多的观察,病人hiv检测结果为阴性,不再有白血病和hiv感染迹象。这一案例的成功,提示ccr5是艾滋病治疗的理想靶点。以辅助受体为靶向的治疗策略为功能性治愈艾滋病提供了一种新思路。已有研究表明,通过基因修饰消除细胞表面ccr5的表达可达到抗hiv病毒感染的目的。技术实现要素:本发明利用crispr技术针对ccr5靶点,设计并筛选获得了安全高效的sgrna,其具有切割效率高,脱靶概率小等优点,在基因编辑效率和使用安全性方面具有最佳效果,并在艾滋病基因治疗的临床研究和应用上具有广泛的前景。所述位点也是基因治疗艾滋病的最佳sgrna位点选择。在一些实施方案中,本文提供针对ccr5基因的sgrna序列,所述sgrna序列选自:ccr5-sgrna1:agggcaactaaatacattct,cr5-sgrna2:tgccaaaaaatcaatgtgaa,ccr5-sgrna3:agtgggactttggaaataca,和ccr5-sgrna4:atgcacagggtggaacaaga。在一些实施方案中,本文提供编码所述sgrna序列的dna序列,例如5'-tcccgttgatttatgtaaga-3',5'-acggttttttagttacactt-3',5'-tcaccctgaaacctttatgt-3',5'-tacgtgtcccaccttgttct-3'。在一些实施方案中,本文提供包含所述sgrna序列或dna序列的载体,如质粒。在一些实施方案中,本文提供包含所述载体的细胞。在一些实施方案中,所述细胞是人细胞、非人哺乳动物细胞、干细胞。在一些实施方案中,本文提供用于在真核细胞中修饰ccr5基因或调节ccr5基因表达的方法,所述方法包括向真核细胞引入所述sgrna序列,所述dna序列和/或所述的载体,并培养所述真核细胞使得sgrna将核酸内切酶定向至ccr5基因。在一些实施方案中,其中所述方法在体外(invitro)和/或离体(exvivo)进行。在一些实施方案中,所述真核细胞是人细胞、非人哺乳动物细胞、干细胞。在一些实施方案中,所述修饰降低ccr5基因表达。在一些实施方案中,所述修饰敲除ccr5基因。在一些实施方案中,本文提供治疗受试者的艾滋病的方法,所述方法包括向受试者引入所述sgrna序列,所述dna序列,所述载体和/或所述修饰的细胞,或向受试者的细胞中引入所述sgrna序列,所述dna序列和/或所述的载体,并培养所述真核细胞使得sgrna将核酸内切酶定向至ccr5基因,从而降低ccr5基因表达,或敲除ccr5基因。在一些实施方案中,其中所述方法在体外和/或离体进行。在一些实施方案中,所述真核细胞是人细胞,例如自体的人细胞或来自其他供体的人细胞。在一些实施方案中,本文提供所述sgrna序列,所述dna序列、所述载体和/或所述细胞在制备用于治疗受试者的艾滋病的药物或试剂盒中的用途。在一些实施方案中,本文提供一种组合物或试剂盒,其包括选自所述sgrna序列,所述dna序列、所述的载体和/或所述的细胞中的任意一项或多项。在一些实施方案中,所述组合物和/或试剂盒包括使用说明书,优选的所述试剂盒可以用于本文描述的用途和方法。在一些实施方案中,所述试剂盒来包括适合用于储存所述sgrna序列、所述载体、和/或所述dna分子的各种试剂如缓冲液等。在一些实施方案中,所述试剂盒包括适合进行所述sgrna序列、所述载体、和/或所述dna分子与目标基因反应的各种试剂包括酶、转化或转染试剂等。在一些实施方案中,所述试剂盒包括适合通过所述sgrna序列、所述载体、和/或所述dna分子通过与目标基因反应而调节目的基因表达(如降低ccr5基因表达的)试剂。在一些实施方案中,本文提供的rna序列如sgrna序列和/或dna序列是分离的序列或合成的序列。附图说明图1:cas9/sgrna体系转染293t细胞荧光照片(左为白光,右为绿色荧光)。图2:4条sgrna对应的t7酶切检测电泳结果。图3:grnat7酶切结果(sgrna1#-sgrna16#)。图4:grnat7酶切结果(sgrna17#-sgrna33#)。具体实施方式现将结合附图更全面地描述本发明,其中描述了本发明的一部分而非全部实施方案。实际上,这些发明可以许多不同的形式来体现,并且不应理解为受本文所示实施方案的限制,其修改形式和其他实施方案也包括在所附权利要求的范围内。1.sgrna一般而言,sgrna是与靶多核苷酸序列具有足够互补性以便与该靶序列杂交并且指导crispr复合物与该靶序列特异性结合的任何多核苷酸序列。在一些实施方案中,可以对本发明的sgrna进行进一步修饰,以使修饰后的sgrna与上文列举的序列ccr5-sgrna1:agggcaactaaatacattct,ccr5-sgrna2:tgccaaaaaatcaatgtgaa,ccr5-sgrna3:agtgggactttggaaataca,和ccr5-sgrna4:atgcacagggtggaacaaga具有约100%,约99%,98%,97%,96%,95%,94%,93%,92%,91%,90%,89%,88%,87%,86%,85%,84%,83%,82%,81%,80%,75%,70%,65%,60%,55%,或50%的同一性,但是仍然能指导crispr复合物与靶序列特异性结合。用于比对序列的算法是本领域已知的,包括clustalw、clustalx、blast等。通过上述序列修饰后,具有所述序列同一性的sgrna序列可以通过本文描述的方法验证其切割效率、脱靶概率,从而获得修饰的序列。在一些实施方案中,具有所述序列同一性的sgrna序列至少具有原序列同样的活性(如切割效率)。在一些实施方案中,具有所述序列同一性的sgrna序列具有提高的活性(如切割效率)。在一些实施方案中,具有所述序列同一性的sgrna序列具有降低的脱靶概率。在一些实施方案中,可以对本发明的sgrna进行进一步修饰,以使修饰后的sgrna与上文列举的序列ccr5-sgrna1:agggcaactaaatacattct,ccr5-sgrna2:tgccaaaaaatcaatgtgaa,ccr5-sgrna3:agtgggactttggaaataca,和ccr5-sgrna4:atgcacagggtggaacaaga具有更短的序列。已经发现能够采用长度短于20nt的sgrna(例如17~18nt的“truncatedsgrna”)而不影响其活性,并可能显著降低脱靶风险。在一些实施方案中,可以采用更短的sgrna,例如17、18、19nt的sgrna。获得更短的sgrna后可以通过本文描述的方法验证其切割效率、脱靶概率,从而获得修饰的序列。在一些实施方案中,所述更短的sgrna序列至少具有原序列同样的活性(如切割效率)。在一些实施方案中,所述更短的sgrna序列具有提高的活性(如切割效率)。在一些实施方案中,所述更短的sgrna序列具有降低的脱靶概率。可以通过适合的测定法来评估sgrna指导crispr复合物与靶序列的序列特异性结合的能力。例如,足以形成crispr复合物的crispr系统的组分,包括待测的sgrna在内,例如通过用编码该crispr序列的组分的载体进行转染而提供到具有相应靶序列的宿主细胞中,随后通过如本文所述方法进行评估。类似地,通过提供该靶序列、包括有待测试的sgrna在内的crispr复合物的组分、和不同于该测试sgrna的对照sgrna,并且比较在该测试sgrna与该对照sgrna反应之间的靶序列处的结合或切割率,可以获得优选的sgrna。在一些实施方案中,还提供了重组模板。重组模板可以是如本文所述的另一个载体的组分,其包含在一个分开的载体中,或者提供为一个分开的多核苷酸。在一些实施方案中,重组模板被设计为用作在同源重组中的模板,如在被作为crispr复合物的一部分的crispr酶切开或切割的靶序列之内或在其附近。在一些实施方案中,该模板多核苷酸与包含靶序列的多核苷酸的一部分互补。在本文中,除另有明确说明外,核苷酸序列的顺序均是指从5'端到3'端的顺序。本文提供的sgrna可以是一种寡核苷酸。术语“寡核苷酸”是指通过共价连接两种以上的核苷酸形成的分子。术语寡核苷酸通常包括寡核苷、寡核苷酸类似物、寡核苷酸模拟物和这些的嵌合组合。当指核苷酸或单体序列时,其可以碱基序列,诸如,例如,a,t(或u),g或c或其类似物的序列。本文中“核苷酸”用在本文中是指包含糖结构部分、碱基结构部分和共价连接的基团(如磷酸或硫代磷酸核苷酸间连接基团)的糖苷,并且包括天然存在的核苷酸(如dna或rna)和包含修饰的糖和/或碱基结构部分的非天然存在的核苷酸,其在本文中还称为“核苷酸类似物”。非天然存在的核苷酸包括具有修饰的糖结构部分(如二环核苷酸或2’修饰的核苷酸,如2’取代的核苷酸)的核苷酸。“核苷酸类似物”是天然存在的核苷酸(如dna或rna核苷酸)利用在糖和/或碱基结构部分中的修饰产生的变体。类似物可以对所述寡核苷酸没有功能性影响或具有功能性影响。例如,通过产生针对靶标的增加的结合亲和力和/或增加的对细胞内核酸酶的抗性和/或增加的转运到细胞内的容易性。在一些实施方案中,所述sgrna包含1个、2个、3个或更多核苷酸类似物。在一些实施方案中,所述寡聚体包含3-8个核苷酸类似物,例如6或7个核苷酸类似物。在一些实施方案中,所述核苷酸类似物包括锁定核酸(lna)。例如,所述核苷酸类似物中1个、2个、3个或更多核苷酸类似物可以是lna。本文定义的修饰的rna和/或dna分子可以含有核苷酸类似物/修饰,例如骨架修饰,糖修饰或碱基修饰。骨架修饰可以是核酸分子中包含的核苷酸骨架的磷酸酯被化学。修饰的修饰骨架的磷酸酯基团可以通过用不同取代基替代一个或多个氧原子来修饰,其实例包括例如硫代磷酸酯。2.crispr酶在一些实施方案中,本发明涉及crispr酶,例如cas9。在一些实施方案中,本发明涉及核酸内切酶如cas9,其包含至少一个核定位信号,至少一个核酸酶结构域,和至少一个与sgrna相互作用以将核酸内切酶靶向用于剪切的特异性核苷酸序列的结构域。在一些实施方案中,所述核酸内切酶经修饰而缺失至少一个功能性核酸酶结构域。在一些实施方案中,本发明涉及分离的核酸,其编码本发明涉及的核酸内切酶。在一些实施方案中,所述核酸针对哺乳动物细胞中的翻译进行密码子优化。在一些实施方案中,所述核酸是针对人细胞中翻译进行密码子优化。在一些实施方案中,编码所述酶的核酸与启动子序列可操作地连接。3.cas9、sgrna载体和递送系统本文提供sgrna载体。sgrna载体包含能被转录为sgrna序列的多核苷酸,所述sgrna序列能对靶基因进行编辑。在一些实施方案中,该载体可以是病毒载体,如慢病毒或杆状病毒或优选地腺病毒/腺病毒相关病毒载体。在一些实施方案中,所述载体包括但不限于,单链、双链、或部分双链的核酸分子,包括dna、rna、或两者的核酸分子。在一些实施方案中,所述载体是质粒。在一些实施方案中,所述载体是病毒载体,例如,逆转录病毒、复制缺陷型逆转录病毒、腺病毒、复制缺陷型腺病毒、以及腺相关病毒(aav))的载体。在一些实施方案中,所述载体包含针对用于表达的宿主细胞而选择的一种或多种调节元件,所述调节元件可操作地连接至待表达的核酸序列如sgrna。所述调节元件包括启动子、增强子、内部核糖体进入位点(ires)、和其他表达控制元件(例如转录终止信号,如多聚腺苷酸化信号和多聚u序列)。在一些实施方案中,本文提供用于sgrna与cas9的双顺反子载体。在一些实施方案中,sgrna与cas9可以由不同启动子启动,例如,cas9由cbh启动子驱动,而sgrna由u6启动子驱动。在一些实施方案中,载体可以被设计为用于在原核或真核细胞中表达crispr转录物(例如核酸转录物、蛋白质、或酶)。在一些实施方案中,使用哺乳动物表达载体,载体能够驱动一种或多种序列在哺乳动物细胞中表达。在一些实施方案中,重组哺乳动物表达载体能够指导核酸优先在特定细胞类型中表达(例如,使用组织特异型调节元件来表达核酸)。组织特异型调节元件是本领域中已知的。在一些实施方案中,将驱动crispr系统的一个或多个元件的表达的一种或多种载体引入到宿主细胞中,使得该crispr系统的这些元件的表达在一个或多个靶位点指导crispr复合物的形成。在一些实施方案中,一个载体包含一个或多个插入位点,可以插入两个或更多个sgrna。在一些实施方案中,本文提供分离或重组体多核苷酸,其包含编码sgrna的rna或dna序列、sgrna载体的各种组分。多核苷酸可以是rna或dna,它们可以是单链或双链,任选地包含合成的、非天然的或改性的核苷酸碱基。dna聚合物形式的多核苷酸可由cdna、基因组dna、合成dna或它们的混合物的一个或多个片段构成。多核苷酸可包括核糖核苷酸以及核糖核苷酸和脱氧核糖核苷酸的组合。所述脱氧核苷酸和核糖核苷酸包括天然存在的分子和合成类似物。本发明所述多核苷酸也涵盖所有形式的序列,包括但不限于单链形式、双链形式、发夹结构、茎环结构等。还提供了包含sgrna载体和其不同组分的重组多核苷酸。重组载体包含人工的或异源的核酸序列的组合例如非天然共存的调控和编码序列。在其它实施方案中,重组载体可包含来源于不同来源的调控序列和编码序列,或来源于相同来源但以不同于天然存在的方式排列的调控序列和编码序列。所述载体可独自使用或与载体组合使用。如果使用载体,则载体的选择取决于用以转化宿主细胞的方法。例如可以使用质粒载体。为了成功地转化、筛选和繁殖包括本发明任何分离核酸片段的宿主细胞,载体上可以包含的遗传元件。因此为了获得显示期望的表达水平和模式的细胞系,可通过dna印迹分析、rna印迹分析、蛋白表达的免疫印迹分析或表型分析等进行筛选。在一些实施方案中,本文所述的一个或多个sgrna载体可以表达盒形式提供在不同细胞类型中表达。所述盒可包括5'和3'调控序列,其被可操作地与本文提供的多核苷酸连接。在转录的5'-3'方向中,所述表达盒可包括转录和翻译起始区(即启动子)、本文所提供的重组多核苷酸、以及转录和翻译终止区(即终止区)。许多启动子可用于本文提供的sgrna载体。通过在sgrna载体中使用不同启动子,可以调节sgrna表达的时间、位置和/或水平。如果需要,sgrna载体可含有启动子调节区(例如赋予诱导型、组成型、环境或发育调节的、或细胞或组织特异性/选择性表达的调节区),转录起始开始位点、核糖体结合位点、rna加工信号、转录终止位点和/或聚腺苷酸化信号。4.导入的方法本文提供的方法包括将sgrna载体导入细胞。本文提供的方法不限于特定方法,只要使多核苷酸进入宿主的至少一个细胞的内部即可。将多核苷酸导入宿主细胞中的方法是本领域已知的,包括但不限于病毒介导的方法。导入包括指将核酸整合进真核或原核细胞中,在该细胞中核酸可整合进细胞的基因组内,并且包括指将核酸或蛋白提供给细胞。转化方案以及用于将多核苷酸序列引入细胞内的方案可以根据被转化细胞的类型而改变。在一些实施方案中,可以使用病毒载体,如慢病毒或杆状病毒或优选地腺病毒/腺病毒相关病毒载体进行导入。在一些实施方案中,可以使用其他递送系统,如酵母系统、微囊泡、基因枪/将载体附接到金纳米粒子上。在所述载体中,sgrna或编码dna可被可操作地连接到一个启动子,而且指导核酸递送到宿主细胞中。5.组合物和/或试剂盒本文提供使用sgrna的方法和提供包含sgrna的组合物和/或试剂盒,所述sgrna当引入细胞时能够对靶基因进行基因编辑。此类方法和组合物包含sgrna载体,所述载体具有编码sgrna的多核苷酸序列。sgrna载体被设计成从所述载体产生sgrna。本文提供的组合物可包含分离或基本上经纯化的多核苷酸。“分离”或“纯化的”多核苷酸,是基本上或本质上不含那些在天然存在环境中正常伴随多核苷酸或与其相互作用的游离组分。因此,分离或经纯化的多核苷酸基本上不含其他细胞物质、或当通过重组体技术生产时的培养基、或基本上不含当通过化学合成时的化学前体或其他化学物质。在一些实施方案中,“分离”多核苷酸不含多核苷酸所来源于的生物的基因组dna中天然地存在于所述多核苷酸两侧(即,位于所述多核苷酸的5'和3'端)的序列。本发明的sgrna、载体、细胞可以用在药物制剂和组合物中,也可制备成方便应用的试剂盒。适当地,所述组合物或试剂盒包含药用溶剂,诸如水或盐水,稀释剂,载体,盐或辅药。本发明还包括含有本发明的寡核苷酸的药物组合物和制剂。本发明的药物组合物可以用于治疗疾病,例如用于基因治疗。在一些实施方案中,本文提供的组合物和/或试剂盒包含:1)编码sgrna的多核苷酸序列,其中该多核苷酸序列包含一种或多种sgrna,该sgrna能够杂交到真核细胞中的靶序列上,2)编码crispr酶的多核苷酸序列。在转录时,该sgrna引导crispr复合物与该靶序列的序列特异性结合,其中该crispr复合物包含与杂交到该靶序列上的该sgrna和crispr酶。在一些实施方案中,所述多核苷酸包含在含有一种或多种载体的载体系统中。6.使用方法在一些实施方案中,本文提供用于在真核细胞中修饰ccr5基因或调节ccr5基因表达的方法,所述方法包括向真核细胞引入所述sgrna序列,所述dna序列和/或所述的载体,并培养所述真核细胞使得sgrna将核酸内切酶定向至ccr5基因。在一些实施方案中,本文提供治疗受试者的艾滋病的方法,所述方法包括向受试者引入所述sgrna序列,所述dna序列,所述的载体和/或所述修饰的细胞,或向受试者的细胞中引入所述sgrna序列,所述dna序列,所述的载体,并培养所述真核细胞使得sgrna将核酸内切酶定向至ccr5基因,从而降低ccr5基因表达,或敲除ccr5基因。在一些实施方案中,本文提供的方法包括:提供1)编码sgrna的多核苷酸序列,其中该多核苷酸序列包含一种或多种sgrna,该sgrna能够杂交到真核细胞中的靶序列上,2)编码crispr酶的多核苷酸序列,该crispr酶任选地包含至少一个或多个核定位序列。在转录时,该sgrna引导crispr复合物与该靶序列的序列特异性结合,其中该crispr复合物包含与杂交到该靶序列上的该sgrna和crispr酶。在一些实施方案中,编码crispr酶的多核苷酸序列是dna或rna。在一些实施方案中,编码crispr酶的多核苷酸序列、sgrna任一者或全部可以是rna。在一些实施方案中,编码crispr酶的序列、sgrna可以是rna并且可以经由脂质体、纳米粒子、微囊泡、或基因枪进行递送。在一些实施方案中,所述多核苷酸被包含在上文描述的一种或多种载体中。在一些实施方案中,本文提供的方法是在体外(invitro)和/或离体(exvivo)地进行的。在一些实施方案中,所述方法包括诱导表达。在一些实施方案中,所述载体是病毒载体,包括aav或慢病毒载体。在一些实施方案中,所述crispr酶是cas9。在一些实施方案中,sgrna的表达是在t7启动子控制下并且是由t7聚合酶的表达驱动的。在一些实施方案中,本文提供的方法包括递送crispr酶,例如通过向细胞递送编码该crispr酶的mrna来进行。在一些实施方案中,本发明的方法包括:1)向真核细胞引入(i)至少一种包含至少一个核定位信号的crispr酶或编码至少一种包含至少一个核定位信号的crispr酶的核酸,(ii)至少一种编码至少一种sgrna的rna或dna,和2)培养所述真核细胞使得sgrna将crispr酶定向至染色体序列中的靶位点,其中所述crispr酶将双链断裂引入该靶位点,并且所述双链断裂通过dna修复过程修复使得所述染色体序列被修饰。在一些实施方案中,所述crispr酶来自cas9。在一些实施方案中,编码所述crispr酶的核酸是mrna。在一些实施方案中,编码所述crispr酶的核酸是dna。在一些实施方案中,所述dna是载体的一部分,所述载体进一步包含编码sgrna的序列。在一些实施方案中,所述真核细胞包括人细胞和非人哺乳动物细胞、干细胞。在一些实施方案中,本文提供的sgrna序列信息如下:ccr5-sgrna1:agggcaactaaatacattct,ccr5-sgrna2:tgccaaaaaatcaatgtgaa,ccr5-sgrna3:agtgggactttggaaataca,ccr5-sgrna4:atgcacagggtggaacaaga。在一些实施方案中,(1)sgrna1通过转入的dna序列(5'-tcccgttgatttatgtaaga-3')进行转录合成,或者直接转入sgrna序列(agggcaactaaatacattct),通过与cas9蛋白结合形成切割复合体,sgrna序列通过识别严格互补序列,从而定位到人类基因组3号染色体ccr5基因序列上(nc_000003.12(46370142..46376206)),对该基因靶序列特定位点完成高效切割。在一些实施方案中,(2)sgrna2通过转入的dna序列(5'-acggttttttagttacactt-3')进行转录合成,或者直接转入sgrna序列(tgccaaaaaatcaatgtgaa),通过与cas9蛋白结合形成切割复合体,sgrna序列通过识别严格互补序列,从而定位到人类基因组3号染色体ccr5基因序列上(nc_000003.12(46370142..46376206)),对该基因靶序列特定位点完成高效切割。在一些实施方案中,(3)sgrna3通过转入的dna序列(5'-tcaccctgaaacctttatgt-3')进行转录合成,或者直接转入sgrna序列(agtgggactttggaaataca),通过与cas9蛋白结合形成切割复合体,sgrna序列通过识别严格互补序列,从而定位到人类基因组3号染色体ccr5基因序列上(nc_000003.12(46370142..46376206)),对该基因靶序列特定位点完成高效切割。在一些实施方案中,(4)sgrna1通过转入的dna序列(5'-tacgtgtcccaccttgttct-3')进行转录合成,或者直接转入sgrna序列(atgcacagggtggaacaaga),通过与cas9蛋白结合形成切割复合体,sgrna序列通过识别严格互补序列,从而定位到人类基因组3号染色体ccr5基因序列上(nc_000003.12(46370142..46376206)),对该基因靶序列特定位点完成高效切割。在一些实施方案中,对于所述sgrna中,所述基因组物种均为人,所述位置均在ccr5基因中,所述的序列信息分别为:agggcaactaaatacattct;tgccaaaaaatcaatgtgaa;agtgggactttggaaataca;atgcacagggtggaacaaga,序列按照5'端到3'端的规则书写。在一些实施方案中,sgrna序列位置信息包括dna序列信息对应的rna序列信息。在一些实施方案中,本文提供用任一所述sgrna序列制备crispr/sgrna载体的方法。在一些实施方案中,所述方法可以包括如下步骤:1)根据所述sgrna序列设计合成crispr体系所需的sgrna载体;2)使用步骤1)获得的载体进行人ccr5基因编辑。在一些实施方案中,所述的crispr/sgrna载体包括dna载体和rna载体。在一些实施方案中,所述方法运用crispr技术进行基因编辑,所需要的靶向引导载体包含本文提供的sgrna序列。本发明的目的之一在于在人ccr5基因上设计出适合crispr技术基因编辑的多个sgrna位点。在一些实施方案中,本文提供sgrna设计方法,所述方法包括:根据对crispr/cas9的sgrna的设计规则,在人ccr5基因的多个外显子上的不同范围内设计多个可能的sgrna。在一些实施方案中,所述方法包括在引物的设计中寻找相应的pam序列。在一些实施方案中,在所需切割位置附近找到pam序列后,将与其相邻的18-23bp的片段可以作为可能的sgrna备选。在一些实施方案中,sgrna的选择位置则分布在ccr5上多个外显子中。本发明的目的之一还在于提供在上述设计的sgrna中筛选出基因编辑安全性高的sgrna位点的方法。在一些实施方案中,所述高安全性sgrna筛选方法包括:在上述多个设计的sgrna的基础上,利用多种预测软件,如crisprdesigntool预测每个sgrna产生脱靶的可能性。不同的软件根据不同的原理预测脱靶。在一些实施方案中,综合考虑多个软件的评分后选择出多个软件中预测脱靶均较低的sgrna。在筛选过程中,以crisprdesigntool为主要参考,我们设定了以下标准:切割效率评判应当在50以上;脱靶位点最高评分不能超过3分;1分以上的脱靶位点不能超过1个;使用cctop工具没有检测出新的脱靶位点。在一些实施方案中,将筛选出的sgrna序列通过引物设计引物的方式连接到载体质粒的相应位置,通过pcr的方式生产质粒进行体内的测试。在一些实施方案中,在构建过程中使用takara公司的primestar酶,使用50ul的反应体系。在引物的设计中将设计好的低脱靶的sgrna的序列加在末端,通过pcr的方式将新的20bp的sgrna序列替换至载体的相应位置上。在一些实施方案中,为方便之后的测试,选用的cas9载体上同时插入了gfp的荧光蛋白序列。转染293t细胞后第二天观察荧光强度(见图1)。通过上述方法,发明人在人ccr5基因外显子上设计了不同sgrna,并对脱靶位点及脱靶概率进行了分析(见下表1-5)。表1.ccr5-sgrna1外显子上可能脱靶位点及脱靶概率得分表2.ccr5-sgrna2外显子上可能脱靶位点及脱靶概率得分sequencescoremismatcheslocustttacattcattttttggtatgg0.63mms[3:9:19]chr17:+62865344ttcccacccattttttggcaggg0.64mms[4:7:8:9]chr1:-184677338ttaatttttattttttggcagag0.54mms[3:5:6:9]chr22:-43219702ttaagattcattttttggtatgg0.34mms[3:5:9:19]chr8:+87153638ctcacagagattttgtggcaggg0.34mms[1:7:8:15]chr14:-95107782ttcacagtgattttttagaaaag0.23mms[7:17:19]chr9:-17299443ttcacatggattttttcacagag0.23mms[8:17:18]chr17:-12901819ttaacattgagattttggcctgg0.24mms[3:11:12:20]chr16:+21664069ttcatatacattttttgccaaag0.24mms[5:8:9:18]chr1:+121312867ttcatatacattttttgccaaag0.24mms[5:8:9:18]chr5:-49692828ttcacttagactctttggcagag0.24mms[6:8:11:13]chr16:+11650266gtgacactgatttcttggcaggg0.24mms[1:3:7:14]chr3:-15298856ttcaccttgtttctttggccaag0.14mms[6:10:13:20]chr1:-192128429tccacatttattgtttggaaagg0.14mms[2:9:13:19]chr2:+170062975ttcagtttcatttttgggcagag0.14mms[5:6:9:16]chr22:+30489822tccacactgatctcttggcatgg0.14mms[2:7:12:14]chr10:+25889757ttcagattaatttttttccagag0.14mms[5:9:17:18]chr4:+129793538ttcacatggattctatggcttgg0.14mms[8:13:15:20]chr2:+210889782tttacattcattttttgatatgg0.14mms[3:9:18:19]chr17:-44617453tttacattcattttttgatatgg0.14mms[3:9:18:19]chr17:-44399599ttcagattgatctttctgcatgg04mms[5:12:16:17]chr4:+81967055ttcacattgatgttttcgtccag04mms[12:17:19:20]chrx:-46457538ttgacattgatcttgaggcagag04mms[3:12:15:16]chr1:-36379536表3.ccr5-sgrna3外显子上可能脱靶位点及脱靶概率得分表4.ccr5-sgrna4外显子上可能脱靶位点及脱靶概率得分sequencescoremismatcheslocusatccacaggaaggaacaagaagg1.33mms[3:10:11]chr19:-43990455agggacagggtggaaaaagagag0.53mms[2:4:16]chr11:-86126410attcacagcctggaacaaaagag0.34mms[3:9:10:19]chr17:+3527515aagctcagggaggagcaagatgg0.24mms[2:5:11:15]chr10:-104374957gtgctcagggtggaaccaaagag0.24mms[1:5:17:19]chr2:-220470931ctggacagggtggaccaagtaag0.24mms[1:4:15:20]chr2:-54159878ctgctcagggtggatccagagag0.24mms[1:5:15:17]chr6:-689106aagcacacggttgaaaaagaagg0.14mms[2:8:12:16]chr3:-37547441ttgcacagtgtgtaacaaaaaag0.14mms[1:9:13:19]chr1:-155889682ctgcagagtgtggaacaggaaag0.14mms[1:6:9:18]chr3:-52873581atgctctggggggaaaaagaaag0.14mms[5:7:11:16]chrx:-41029225acgcaccgggtggaagtagatgg0.14mms[2:7:16:17]chr22:+50688848atgaagagggtgaaacaggaaag0.14mms[4:6:13:18]chr1:+233397754atgcccagagtgtaacacgatgg0.14mms[5:9:13:18]chr1:+186113596attcacagggaggcacaggacag04mms[3:11:14:18]chr6:-166354166atgcacaggctggcaccggaagg04mms[10:14:17:18]chr9:+139903922表5.低脱靶grna筛选在一些实施方案中,本发明的目的之一在于提供在以上这些sgrna中进一步筛选出具有高效基因编辑的sgrna位点的方法。所述方法包括获得一系列低脱靶的sgrna,连接到相应载体上,检测这些sgrna的切割效率。此前有相关文章报道过crispr/cas9的切割效率很大程度上受到了sgrna的影响。在一些实施方案中,应用了293t细胞进行了测试了上述筛选后的高安全性的sgrna。在一些实施方案中,所述的高效基因编辑的sgrna筛选方法如下:在获得了各个sgrna的荧光数据后,进一步检测了每种sgrna的切割效率。将转染培养了的293t细胞收集后,使用qiagen盒子提取基因组。使用不同sgrna在ccr5的不同位置上通过pcr扩增包含了预期切割位点的序列。在使用tiangen试剂盒回收pcr产物之后,使用neb的t7限制性内切酶切割。这一酶会非特异性切割dna上不完美吻合的位置。如果在crispr/cas9作用下,目标位置有可能被切割,然后再nhej的作用下会产生能被t7识别的不匹配序列,从而被切割。如图2。通过上述检测方法,发明人测试的33组grna切割效率相关数据如下表6:表6:grna切割效率由上表可以看出12#(grna1),17#(grna2),9#(grna3),19#(grna4)四组grna切割效率最高(t7酶切结果见图3,图4),均高于70%的切割效率。在切割过程中,可以使用了20ul的体系,其中包括了2ul10xnebbuffer2,1000ngdna,以及补足体系的去离子水。为了保证酶切的效果,在加入t7酶之前,pcr产物经过了退火程序。然后在t7酶切1个小时以后进行琼脂糖电泳鉴定。为了进一步确定所选sgrna的切割效率,对有清晰t7切割的pcr产物,将其连接在transgene公司生产的bluntsimple载体上进行测序。并挑出30个至100个克隆进行测序,确定切割比例及切割方式。通过本发明的方法,综合比对了细胞死亡率,核转效率,以及切割效率之后,能够获得高效的、低脱靶效率的sgrna。7.应用本发明的寡核苷酸可以用作研究试剂,例如,用于诊断、治疗和预防。在研究中,所述寡核苷酸可以用于特异性结合目的,可以对所述基因进行编辑,例如降低其表达或将其敲除,由此促进对靶标的功能性分析或对其作为治疗干预的靶标的评估。对于治疗剂,患有艾滋病的受试者可以通过施用本发明所述的寡核苷酸进行治疗。进一步提供治疗怀疑患有或倾向于患有艾滋病的哺乳动物(如治疗人)的方法,所述通过通过施用治疗或预防有效量的一种或多种本发明的寡核苷酸或组合物。本发明所述的寡核苷酸或药物组合物典型地以有效量施用。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。1.sgrna质粒载体的构建(1)引物设计与合成我们从人ccr5基因第一个外显子开始到ccr5-δ32位置为止,参照sgrna设计规则找到正、反义链上所有的sgrna序列设计引物,通过生物信息学评价,筛选低脱靶的sgrna设计相应的引物,发送到引物合成公司进行合成。(2)sgrna引物溶解及退火取公司合成引物,3000rpm离心5min,加te溶解至200um,37℃放置15min。取5μl的forwardprimer及对应的reverseprimer于pcr管中,混匀。退火程序:95℃x10min95℃x1s(-2.0℃/cycle),goto2,4times85℃x1s(-0.2℃/cycle),goto4,299times25℃x1min(3)sgrna插入片段与载体的连接1)去30ug插入片段与5ug的sgrna载体配成连接混合液(solutionⅰ体系),37℃连接1小时。2)使用全式金trans5α或者t1感受态菌,向每一管中加入25μl感受态菌液,冰上放置30min。3)42℃水浴90s,之后冰上复苏3min,加入100μl无抗性lb培养基。37℃摇床中复苏15min-30min。4)将125μl菌液中65μl涂于0.001%amp的lb平板上,放入37℃培养箱中培养12h-16h。5)每个平板挑取4个克隆,溶于100μlamplb培养基中。37℃摇床培养3-10h。测序鉴定出正确的克隆。2.sgrna质粒载体的高度纯化该载体的高度纯化,采用qiagen去内毒素套装的中提试剂盒,其每次提取dna总量可在50-100μg。(1)提取talen质粒需要的菌液准备1)配置100ml0.001%amp的lb培养基,将菌种中50μl放入锥形瓶中,在37℃摇床中摇16h。2)取出多个50ml离心管,将菌液分三次倒入50ml离心管中,12000rpm/min离心7min,倒掉上清液。(2)qiagen试剂盒中提过程1)向每管中加入4ml加入好rnasea的p1溶液,重悬菌液。2)加入4mlp2溶液,颠倒混匀数次,室温放置3-5分钟,此时溶液会出现粘稠状物质并呈现蓝色。3)加入10mlp3溶液,溶液中蓝色退去,出现白色絮状沉淀,颠倒混匀数次使得其均匀,放入4℃离心机20000xg离心30分钟,将上清液转移到一个新的50ml管中,注意尽量不要倒出白色沉淀物。4)向每个50ml离心管中加入840μlendotoxicremovalbuffer,冰上放置30min。5)使用4mlbufferqbt平衡qiagen-tip100吸附柱。6)将含有质粒的混合液通过注射器与滤膜,将液体注入到吸附柱中。7)使用10ml的qcbuffer洗涤qiagen-tip100吸附柱两次。8)将吸附柱架在一个新的离心管上,用5ml的bufferqf洗脱dna。9)向新的离心管中加入3.5ml异丙醇,放入4℃离心机20000xg,离心30分钟,去除上清液。10)向新的离心管中加入2ml70%乙醇洗涤,放入4℃离心机20000xg,离心30分钟。11)转移至细胞间,去除上清液,晾干,用40-100μl无菌水或te洗脱液溶解质粒,放置5min。12)分装,使用nanodrop测定质粒溶液的吸光度和浓度,一般而言a260/a280大于1.9,a260/a230接近或大于2,浓度大于500ng/μl可进行下一步实验。3.sgrna质粒阳转与纯化(1)sgrna质粒的阳性转化1)使用全式金trans5α或者t1感受态菌,向每一管中加入25μl感受态菌液,冰上放置30min。2)42℃水浴90s,之后冰上复苏3min,加入100μl无抗性lb培养基。37℃摇床中复苏15min-30min。3)将125μl菌液中65μl涂于0.001%amp的lb平板上,放入37℃培养箱中培养12h-16h。4)每个平板挑取2个克隆,溶于100μlamplb培养基中。37℃摇床培养3-10h。(2)sgrna质粒的提取按照unitassembling质粒小提方式相同的提取方式,保种并提取阳性转化后摇起来的感受态大肠杆菌中的质粒。4.在293t细胞上进行sgrna效率筛选293t细胞易于存活,生长速度快,基因组状态较为松弛,对于细菌内毒素抗性显著强于人类cd34+细胞群。适合用于该检测实验。转染细胞之前的质粒要进行灭菌处理,尽量消除其污染细胞的可能性。(1)质粒的沉降和灭菌1)将纯化好sgrna质粒载体中加入两倍体积无水乙醇和1/10体积的3m醋酸钠,混合均匀,放入-20℃中沉降30min以上。2)4℃12000rpm/min离心15min,此时可见白色沉淀团块,除去上清液,加入1ml70%乙醇洗涤。3)4℃12000rpm/min离心15min,转移至细胞间超净台,管壁喷洒酒精。4)移走70%乙醇,吹干残留液体,加入10-20μl无菌水溶解5-10min。5)转移出1μl至一个新的小管中,测浓度和a260/a280,a260/a230,要求达到浓度>300ng/μl,a260/a280>1.9,a260/a230接近或>2.0。(2)脂质体转染sgrna和cas9质粒进入293t细胞1)观察293t细胞贴壁情况,对于生长均匀,无重叠,且覆盖培养皿70-80%面积的293t细胞适宜进行转染实验。2)按照孔板和细胞数量的大小,配制ltx混合液和质粒-plus混合液,按照invitrogen公司提供的表进行配制(表7,经修改)。通常,按照1×106细胞转染单侧1.5μg或2μgtalen(共4μg),转染2μg。3)将混合好的dna-plus混合液缓慢加入ltx混合液中,迅速振荡混匀。4)混合好所有的管后再次使用vortex振荡,将混合液缓慢加入铺有293t细胞的孔中,在镜下观察细胞状态和混合的颗粒大小及均匀程度。5)放在37℃二氧化碳培养箱中,晃动均匀,培养。6)12h后观察gfp质粒孔的状态,判断转染效率。更换培养基,将细胞重新放回37℃二氧化碳培养箱中,至第72小时。(3)提取293t细胞基因组使用tiangen细胞基因组提取试剂盒,提取293t细胞基因组。1)293t细胞预处理:吸走293t细胞上层的培养基成分,使用胰蛋白酶适量(1ml对应6孔板,500μl对应12孔板),在37℃温箱中消化3min。2)从细胞间将孔板转移到分子实验区,将孔内细胞转移到1.5mltube中,12000rpm/min离心2min,去上清。3)用1mlpbs洗涤孔板,并将洗涤液转移到tube中,12000rpm/min离心2min,去上清。4)按照200μlga,20μl蛋白酶k(试剂盒自带),4μlrnasea(试剂盒自带)配置混合重悬液,加入到tube中重悬细胞团至没有团块。5)加入200μlgb,混匀,70℃放置15min,每隔5min颠倒混匀一次,至溶液清亮。6)加入200μl无水乙醇,短暂离心除去管壁水珠。7)将混合液加入到已插入到收集管内的cb3柱中,12000rpm/min离心30s,倒掉收集管内的废液。8)将600μlgd加入到cb3柱中,12000rpm/min离心30s,倒掉收集管内的废液。9)将700μlpw加入到cb3柱中,12000rpm/min离心30s,倒掉收集管内的废液。10)重复9)。11)12000rpm/min离心2min,甩干pw以免其对后续反应产生影响,将cb3柱插入到一个干净的1.5mltube内,室温放置5min。12)将洗脱液te放于65℃空气浴上加热。13)向cb3柱中央加入35μl洗脱缓冲液te,12000rpm/min离心30s。14)将1.5mltube中的洗脱缓冲液再次加入cb3柱中,12000rpm/min离心30s。15)使用nanodrop测定质粒溶液的吸光度和浓度,一般而言a260/a280大于1.8,浓度大于10ng/μl可进行下一步实验。16)抽取5μl左右载体进行1%琼脂糖凝胶电泳鉴定,有单一条带且其大小在15000bp左右或以上的可进行下一步实验。(4)pcr反应获得特异性切割部分附近的片段设计引物,使得pcr产物的大小在500-1500之间,同时可以覆盖sgrna所覆盖的序列范围,引物距离sgrna切割位点各有200bp以上的距离。由于有些sgrna作用的位点在基因组上位置比较集中,故可以共用引物,则引物数量比sgrna数量显著减少,表8列出了部分最常用的检测引物及其可以检测的sgrna(由上海生工公司合成)。顺序大体代表引物常用程度。pcr反应采用takaraprimestar体系如下:总pcr体系大小:50μl(一般为两管)在pcr仪中使用如下pcr程序进行反应:98℃5min,98℃10s,58℃15s,72℃50s)循环35次;72℃10min;4℃forever。1)使用1%琼脂糖凝胶电泳,判断条带是否单一及有无正确大小的条带。若条带单一且大小正确,采用pcr产物电泳回收试剂盒;若条带有大小正确但不单一,则采用琼脂糖凝胶回收试剂盒。2)对于回收的片段使用nanodrop检测其浓度和吸光度,一般而a260/a280大于1.8,浓度大于20ng/μl可进行下一步实验。(5)t7内切酶i检测突变片段t7内切酶i可以识别双链dna颈环或泡状结构等不完全匹配的部分,并将其切成两部分。由于cas9/sgrna作用会引发基因组非同源末端修复机制并产生不同的随机修复片段,当混合有各种处理过的片段的pcr产物在一起进行梯度退火时,片段经历不断的“解链-结合”的过程,使得两端匹配但cas9/sgrna作用区不匹配的片段比例增加,而这种片段正可以被t7内切酶i所切割,产生两条条带。1)将pcr产物与nebbuffer2及水按照以下的配方混合:h2ouptobuffer22μlpcr500μ放入pcr仪中,以如下程序进行多重梯度退火:95℃5min;95℃-85℃,每3秒钟下降2℃;85℃1min;85℃-25℃,每秒钟下降0.3℃,每逢5x℃停留1min;25℃forever。2)向处理过的pcr产物混合液中加入1μlt7endonuclease,放入37℃。pcr仪中温浴2h。3)配置2%的琼脂糖凝胶,向pcr产物中加入takara10×loading。4)输出电泳图,判断cas9/sgrna在293t细胞系上的效率特性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1