联薁二酰亚胺衍生物、其制备方法和应用与流程
本发明涉及一种联薁二酰亚胺衍生物、其制备方法和应用。
背景技术:
有机薄膜晶体管(otft)是通过改变电场来控制半导体材料导电能力的有源器件。与无机薄膜晶体管相比,有机薄膜晶体管的成膜技术更多、器件的制作工艺较为简单、能够有效地降低器件的成本,可广泛应用于电子标签(rfid)、有源矩阵显示、有机传感器、存储器、电子纸等领域(arias,a.c.etal.chem.rev.2010,110,3;huitema,h.e.a.etal.nature2001,414,599;rogers,j.a.etal.science2010,327,1603.;specialissue:organicelectronicsandoptoelectronics,forrest,s.r.;thompson,m.e.ed.chem.rev.2007,107,923;gelinck,g.etal.adv.mater.2010,22,3778等)。随着otft和相关领域技术的发展,otft将在最具增长潜力的柔性显示驱动、有机电子标签、有机传感器等方面发挥举足轻重的作用,一些轻薄、便携、可弯曲、可穿戴、个性时尚的有机电子产品将逐步走进人们的生活,并将给电子产业和人类的生活带来革命性的变化。
有机半导体材料是有机场效应晶体管的重要组成部分,起载流子产生、注入和传输的作用。按其传输载流子的类型,分为p-型(传输空穴)、n-型(传输电子)和双极性(既传输空穴也传输电子),材料的内在性质决定了载流子迁移的快慢和稳定性,从而决定了器件的性能。因此,合成和开发高性能的有机半导体材料,是制备otft器件的关键。近年来,芳香二酰亚胺及其衍生物由于其较低的lumo能级,较高的热稳定性和化学稳定性以及可调控的分子自组装行为等特点,引起了人们的广泛重视(lei,t.;dou,j.h.;cao,x.y.;wang,j.y.;pei,j.j.am.chem.soc.2013,135,12168.)。在共轭π-骨架引入酰亚胺基团,一方面可以降低分子的lumo能级,另一方面会增加分子内电子云的极化程度造成分子间的静电吸引,从而使分子排列更为紧密。此外,还可以通过酰亚胺氮端的取代基对分子自组装行为进行有效的调控,从而获得高性能的半导体材料(尤其是n-型半导体材料)。从这些芳香酰亚胺出发来合成新的聚合物或小分子n-型材料,已经成为目前n-型半导体材料发展的一条主要途径。
在化石能源日益枯竭、环境污染日渐严重的今天,开发将太阳能转换成电能的太阳能电池是解决人类面临的能源和环境问题的根本途径。传统的无机半导体太阳能电池,如单晶硅电池,虽然具有高达20%的转化效率和长达25年的工作寿命,但其生产工艺复杂、成本高、制作过程耗能高并且会造成二次污染;其转换效率也基本达到极限值,使得此类电池的进一步改进受到相当大的限制。有机光伏器件因具有重量轻、成本低以及可以制成柔性大面积器件等突出优点,成为近年来太阳能电池的研究热点(段晓飞,王金亮,毛景,裴坚。有机太阳能电池材料的研究进展。大学化学,2005,20(3):1-9;黎立桂,鲁广昊,杨小牛,周恩乐。聚合物太阳能电池研究进展。科学通报,2006,51(21):2457-2468;brabec,c.j.;sariciftci,n.s.;hummelen,j.c.adv.funct.mater.2001,11,15.)。然而,相对于无机太阳能电池,有机太阳能电池效率较低,性能也不够稳定。为了提高其能量转换效率,国内外化学家、物理学家和材料学家分别从材料和器件的角度对有机太阳能电池进行了深入研究。研究发现,作为有机太阳能电池的重要组成部分,光活性层材料的改进对于提高有机太阳能电池的性能具有非常重要的意义(liang,y.y.etal.adv.mater.2010,22,e135-e138.)。
薁是一种青蓝色的具有特殊分子结构的非苯芳香化合物,是萘的同分异构体。从分子结构上看,薁是一个由带正电荷的环庚三烯和带负电荷的环戊二烯并合而成的具有较大分子偶极矩(1.08d)的化合物。近几年,薁类化合物在有机电子和光伏器件,包括有机场效应晶体管(ofet)、有机太阳能电池(opv)和钙钛矿太阳能电池等领域的研究使该类化合物进一步引起了人们的关注和兴趣(yamaguchi,y.etal.j.am.chem.soc.2013,135,19095;yao,j.j.etal.macromolecules2015,48,2039;puodziukynaite,e.etal.j.am.chem.soc.2014,136,11043;nishimura,h.etal.j.am.chem.soc.2015,137,15656.)。因此,为了寻求结构多样、能级可调控的新型芳香酰亚胺类化合物,同时进一步探究薁类化合物特殊的物理化学性质,发明人披露了一种未见报道的联薁二酰亚胺衍生物及其合成,并将其应用于otft和opv器件。
技术实现要素:
本发明所要解决的技术问题是为了寻求结构多样、能级可控的新型芳香酰亚胺类化合物,同时进一步探究薁类化合物特殊的物理化学性质,而提供了一种联薁二酰亚胺衍生物、其制备方法和应用。本发明的联薁二酰亚胺衍生物具有不同的能级、大π共轭体系和柔性促溶的烷基链,可以用溶液加工的方法低成本制备有机电子器件(如otft、opv等);同时本发明的联薁二酰亚胺衍生物制备方法简单,原料易于合成,成本低,得到的目标化合物的纯度高。
本发明提供了一种通式i所示的联薁二酰亚胺衍生物:
其中,x1和x2独立地为h、卤素、取代或未取代的c6-c20芳基或取代或未取代的c2-c20杂芳基;所述的取代的c6-c20芳基或所述的取代的c2-c20杂芳基中的取代基为下列基团中的一个或多个(优选1-6个,更优选1-3个):卤素、取代或未取代的c1-c4烷基或取代或未取代的c6-c10芳基;所述的取代的c1-c4烷基中取代基是指被一个或多个(优选1-6个,更优选1-3个)卤素所取代:所述的取代的c6-c10芳基中的取代基是指被下列基团中的一个或多个(优选1-6个,更优选1-3个)所取代:卤素、c1-c4烷基或卤素取代的c1-c4烷基;
r1和r2独立地为h、取代或未取代的c1-c48烷基、取代或未取代的c2-c48烯基或取代或未取代的c6-c24环烷基;其中,所述的取代的c1-c48烷基、取代的c2-c48烯基或取代的c6-c24环烷基中的取代基是指被下列基团中的一个或多个(优选1-6个,更优选1-3个)所取代:卤素、c1-c4烷基、c1-c4硅烷基或卤素取代的c1-c4烷基。
x1和x2中,所述的卤素或所述的卤素取代的c1-c4烷基中所述的卤素优选f、cl、br或i。
x1和x2中,所述的c1-c4烷基、所述的取代或未取代的c1-c4烷基中所述的c1-c4烷基或所述的卤素取代的c1-c4烷基中所述的c1-c4烷基优选甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基。
x1和x2中,所述的卤素取代的c1-c4烷基优选被一个或多个(优选1-6个,更优选1-3个)卤素所取代的c1-c4烷基。所述的被一个或多个卤素所取代的c1-c4烷基优选三氟甲基。
x1和x2中,所述的取代或未取代的c6-c20芳基优选取代或未取代的c6-c10芳基。所述的取代或未取代的c6-c10芳基优选取代或未取代的苯基(例如
x1和x2中,所述的取代或未取代的c2-c20杂芳基优选杂原子选自o、n、s和se,杂原子数为1-4个的取代或未取代的c2-c10杂芳基。所述的取代或未取代的c2-c10杂芳基优选取代或未取代的吡啶基
r1和r2中,所述的取代或未取代的c1-c48烷基优选取代或未取代的c6-c20烷基。所述的取代或未取代的c6-c20烷基优选取代或未取代的c8-c16烷基。所述的取代或未取代的c8-c16烷基优选取代或未取代的正辛基或取代或未取代的2-己基-癸烷基。
r1和r2中,所述的取代或未取代的c2-c48烯基优选取代或未取代的c2-c20烯基。
r1和r2中,所述的取代或未取代的c6-c24环烷基优选取代或未取代的c6-c20环烷基。
在本法明一优选实施方式中,通式i所示的联薁二酰亚胺衍生物中,x1和x2相同;r1和r2相同。
本发明中,所述的通式i所示的联薁二酰亚胺衍生物,其优选下列任一化合物:
本发明还提供了一种通式i所示的联薁二酰亚胺衍生物的制备方法,其包含下列任一方法:
方法一包括下列步骤:将通式i-1所示的化合物与酰氯化试剂进行如下所示的反应,制得通式i所示的联薁二酰亚胺衍生物;
方法二包括下列步骤:在碱的作用下,将通式i-1所示的化合物与酸酐进行如下所示的反应,制得通式i所示的联薁二酰亚胺衍生物;
其中,x1、x2、r1和r2的定义均同前所述。
方法一中,所述的酰氯化试剂可为有机合成领域此类反应常规的酰氯化试剂,优选二氯亚砜、三氯氧磷、五氯氧膦和草酰氯中的一种或多种。当所述的酰氯化试剂为液体时,所述的酰氯化试剂既作为反应物又作为反应溶剂。所述的酰氯化试剂的用量可不作具体限定,只要不影响反应的进行即可,当所述的酰氯化试剂为液体时,所述的酰氯化试剂与通式i-1所示的化合物的体积质量比优选20ml/g-100ml/g,优选40ml/g-60ml/g(例如55.6ml/g)。当所述的酰氯化试剂为固体时,所述的酰氯化试剂与通式i-1所示的化合物的摩尔比优选0.1:1-20:1,更优选1:1-10:1,最优选1:1-5:1。当所述的酰氯化试剂为固体时,所述的反应优选在溶剂存在下进行,所述的溶剂和用量可为有机合成领域此类反应常规的溶剂和用量,只要不影响反应的进行即可。所述的反应的温度可为有机合成领域此类反应常规的温度,优选酰氯化试剂或溶剂常压下回流温度,例如60-110℃,更优选75-85℃。所述的反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-1所示的化合物消失时作为反应的终点。所述的反应的时间优选1-5小时,更优选2-3小时。
方法二中,所述的酸酐可为有机合成领域此类反应常规的酸酐,优选乙酸酐、邻苯二甲酸酐和马来酸酐中的一种或多种。所述的碱可为有机合成领域此类反应常规的碱,优选无机碱。所述的无机碱优选乙酸钠。所述的碱的用量可不作具体限定,只要不影响反应进行即可,其与通式i-1所示的化合物的摩尔比优选5:1-20:1,更优选10:1。当所述的酸酐为液体时,所述的酸酐既作为反应物又作为反应溶剂。当所述的酸酐为液体时,所述的酸酐与通式i-1所示的化合物的体积质量比优选50ml/g-200ml/g,优选100ml/g-160ml/g(例如153.8ml/g)。当所述的酸酐为固体时,所述的酸酐与通式i-1所示的化合物的摩尔比优选0.1:1-20:1,更优选1:1-10:1,最优选1:1-5:1。当所述的酸酐为固体时,所述的反应优选在溶剂存在下进行,所述的溶剂和用量可为有机合成领域此类反应常规的溶剂和用量,只要不影响反应的进行即可。所述的反应的温度可为有机合成领域此类反应常规的温度,优选酸酐或溶剂常压下回流温度,例如135-145℃。所述的反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-1所示的化合物消失时作为反应的终点。所述的反应的时间优选1-10小时,更优选4-8小时。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:溶剂中,将通式i-2所示的化合物与通式i-3所示的化合物(r1nh2和/或r2nh2)进行如下所示的酰胺化反应,制得所述的通式i-1所示的化合物;
其中,x1、x2、r1和r2的定义均同前所述。
所述的通式i-1所示的化合物的制备方法中,所述的溶剂可为有机合成领域此类反应常规的溶剂,优选卤代烃类溶剂。所述的卤代烃类溶剂优选二氯甲烷。所述的溶剂的量可不作具体限定,只要不影响反应进行即可。所述的通式i-2所示的化合物与通式i-3所示的化合物的用量可不作具体限定,为有机合成领域此类反应常规的用量,通式i-2所示的化合物与r1nh2和/或r2nh2的摩尔比优选为1:1-1:5,更优选1:2-1:4。所述的酰胺化反应的温度可为有机合成领域此类反应常规的温度,优选35-100℃,更优选60-80℃。所述的酰胺化反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-1所示的化合物消失时作为反应的终点。所述的反应的时间优选1-12小时,更优选2-4小时。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:在脱水剂的作用下,将通式i-4所示的化合物进行如下所示的脱水反应,制得所述的通式i-2所示的化合物;
其中,x1和x2的定义均同前所述。
所述的通式i-2所示的化合物的制备方法中,所述的脱水剂可为有机合成领域此类反应常规的脱水剂,优选乙酸酐和/或五氧化二磷。当所述的脱水剂为液体时,所述的脱水剂既作为反应物又作为反应溶剂。当所述的脱水剂为液体时,所述的脱水剂与通式i-4所示的化合物的体积质量比优选5ml/g-100ml/g,优选10ml/g-30ml/g(例如24.4ml/g)。当所述的脱水剂为固体时,所述的脱水剂与通式i-4所示的化合物的摩尔比优选0.1:1-20:1,更优选1:1-10:1,最优选1:1-5:1。当所述的脱水剂为固体时,所述的反应优选在溶剂存在下进行,所述的溶剂和用量可为有机合成领域此类反应常规的溶剂和用量,只要不影响反应的进行即可。所述的脱水反应的温度可为有机合成领域此类反应常规的温度,优选脱水剂或溶剂常压下回流温度,例如135-145℃。所述的脱水反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-4所示的化合物消失时作为反应的终点。所述的脱水反应的时间优选1-10小时,更优选2-4小时。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:溶剂中,在碱的作用下,将通式i-5所示的化合物进行如下所示的水解反应,制得所述的通式i-4所示的化合物;
其中,ra、rb、rc和rd独立地为c1-c4烷基(所述的c1-c4烷基优选甲基、乙基、正丙基、异丁基、正丁基、异丁基或叔丁基);x1和x2的定义均同前所述。在本发明一优选实施方式中,ra、rb、rc和rd相同,优选乙基。
所述的通式i-4所示的化合物的制备方法中,所述的溶剂可为有机合成领域此类反应常规的溶剂,优选水和/或有机溶剂。当所述的溶剂为水和有机溶剂的混合溶剂时,水和有机溶剂的用量可不作具体限定,只要不影响反应进行即可,水和有机溶剂的体积比优选1:10-1:20,例如1:16。所述的有机溶剂优选c1-c4醇类溶剂和/或醚类溶剂。所述的c1-c4醇类溶剂优选乙醇。所述的醚类溶剂优选四氢呋喃。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-5所示的化合物的体积质量比优选10ml/g-100ml/g,优选30ml/g-60ml/g(例如40-55ml/g)。所述的碱可为有机合成领域此类反应常规的碱,优选无机碱。所述的无机碱优选氢氧化钾、氢氧化钠和氢氧化锂中的一种或多种。所述的碱的用量可不作具体限定,只要不影响水解反应进行,即可,其与通式i-5所示的化合物的摩尔比优选10:1-30:1,更优选20:1。所述的水解反应的温度可为有机合成领域此类反应常规的温度,优选常压下溶剂回流温度,例如60-110℃。所述的水解反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-5所示的化合物消失时作为反应的终点。所述的水解反应的时间优选5-36小时,更优选10-24小时。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:气体保护和无水条件下,避光条件下,溶剂中,在镍催化剂的作用下,将通式i-6所示的化合物和通式i-6a所示的化合物进行如下所示的偶联反应,制得所述的通式i-5所示的化合物;
其中,ra1和ra2独立地为卤素(例如cl、br或i);ra、rb、rc、rd、x1和x2的定义均同前所述。在本发明一优选实施方式中,ra1和ra2相同,优选cl。
所述的通式i-5所示的化合物的制备方法中,所述的气体保护中的气体优选氮气。所述的镍催化剂可为有机合成领域此类反应常规的镍催化剂,优选由氯化镍、溴化镍和碘化镍中的一种或多种,在锌粉和有机合成领域常规的磷配体等还原剂还原下制备得到的零价镍催化剂,或者有机合成领域其他常规法方法制得的零价镍催化剂,优选ni(cod)2(二环辛二烯合镍)。所述的镍催化剂的用量可不作具体限定,为有机合成领域此类反应常规的用量,其与通式i-6所示的化合物的摩尔比优选0.55:1-1:1,更优选0.55:1-0.8:1。所述的溶剂可为有机合成领域此类反应常规的溶剂,优选酰胺类溶剂。所述的酰胺类溶剂优选n,n-二甲基甲酰胺(dmf,例如重蒸dmf)。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-6所示的化合物的体积质量比优选1ml/g-50ml/g,优选5ml/g-25ml/g。所述的偶联反应的温度可为有机合成领域此类反应常规的温度,优选40-60℃,例如50℃。所述的偶联反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-6所示的化合物消失时作为反应的终点。所述的偶联反应的时间优选2-10小时,更优选5-6小时。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:溶剂中,在亚硝酸类化合物的作用下,将通式i-7所示的化合物和氯化试剂进行如下所示的氯化反应,制得所述的通式i-6或i-6a所示的化合物;
或者,溶剂中,气体保护和无氧条件下,在碱和钯催化剂的作用下,将通式i-8所示的化合物和通式i-9所示的化合物(x1-br或x2-br)进行如下所示的偶联反应,制得所述的通式i-6或i-6a所示的化合物;
其中,rax和rbx分别为ra和rb,或者rax和rbx分别为rc和rd;rcx为硼保护基(例如
所述的通式i-6或i-6a所示的化合物的制备方法中,所述的氯化反应的条件可为有机合成领域此类反应常规的条件。其中,所述的亚硝酸类化合物可为有机合成领域此类反应常规的亚硝酸类化合物,优选亚硝酸异戊酯。所述的亚硝酸类化合物的用量可不作具体限定,为有机合成领域此类反应常规用量,其与通式i-7所示的化合物的摩尔比优选1:1-10:1,更优选2:1-5:1(例如3:1)。所述的氯化试剂可为有机合成领域此类反应常规的氯化试剂,优选三甲基氯硅烷或氯化氢气体。所述的氯化试剂的用量可不作具体限定,为有机合成领域此类反应常规用量,其与通式i-7所示的化合物的摩尔比优选1:1-10:1,更优选2.5:1-5:1(例如3:1)。所述的溶剂可为有机合成领域此类反应常规的溶剂,优选卤代烃类溶剂。所述的卤代烃类溶剂优选氯仿。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-7所示的化合物的体积质量比优选1ml/g-100ml/g,优选3ml/g-50ml/g(例如40.5ml/g)。所述的氯化反应的温度可为有机合成领域此类反应常规的温度,优选室温,例如10-40℃。所述的氯化反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-7所示的化合物消失时作为反应的终点。所述的氯化反应的时间优选2-24小时,更优选10-12小时。
所述的通式i-6或i-6a所示的化合物的制备方法中,所述的偶联反应的条件可为有机合成领域此类反应常规的条件。其中,所述的气体保护中的气体优选氮气。所述的碱可为有机合成领域此类反应常规的碱,优选无机碱。所述的无机碱优选乙酸钾和/或碳酸氢钠。所述的碱的用量可为有机合成领域此类反应常规的用量,其与通式i-8所示的化合物的摩尔比优选1:1-5:1,更优选2:1-4:1。所述的钯催化剂可为有机合成领域此类反应常规的钯催化剂,优选pd(pph3)2cl2(二氯二(三苯基磷)合钯)和/或pd(pph3)4(四(三苯基膦))。所述的钯催化剂的用量可为有机合成领域此类反应常规的用量,其与通式i-8所示的化合物的摩尔比优选0.01:1-5:1,更优选0.05:1-1.65:1;最优选0.2:1-0.5:1。通式i-8所示的化合物和通式i-9所示的化合物的用量关系可不作具体限定,只要不影响反应进行即可,二者摩尔比优选0.5:1-1:1.5,例如1:1.2。所述的溶剂可为有机合成领域此类反应常规的溶剂,优选水和/或有机溶剂。当所述的溶剂为水和有机溶剂的混合溶剂时,水和有机溶剂的用量可不作具体限定,只要不影响反应进行即可,水和有机溶剂的体积比优选1:3-1:5。所述的溶剂优选除氧处理。所述的有机溶剂优选c1-c4醇类溶剂、芳烃类溶剂和醚类溶剂中的一种或多种。所述的c1-c4醇类溶剂优选乙醇。所述的芳烃类溶剂优选甲苯。所述的醚类溶剂优选1,4-二氧六环。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-8所示的化合物的体积质量比优选1ml/g-50ml/g,优选5ml/g-15ml/g。所述的偶联反应的温度可为有机合成领域此类反应常规的温度,一般为常压下溶剂回流温度,优选40-100℃,例如60-100℃。所述的偶联反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-8所示的化合物消失时作为反应的终点。所述的偶联反应的时间优选30分钟-10小时,更优选2-9小时(例如5-6小时)。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:溶剂中,气体保护和无氧条件下,在碱和钯催化剂的作用下,将通式i-10所示的化合物和通式i-11所示的化合物(x-rex)进行如下所示的偶联反应,制得所述的通式i-7所示的化合物;
其中,rfx优选卤素(例如f、cl、br或i)或-otf(对三氟甲磺酸酯);rex优选-b(oh)2、硼酸酯(例如二氧硼戊环
所述的通式i-7所示的化合物的制备方法中,所述的偶联反应的条件可为有机合成领域此类反应常规的条件。其中,所述的气体保护中的气体优选氮气。所述的碱可为有机合成领域此类反应常规的碱,优选无机碱。所述的无机碱优选乙酸钾和/或碳酸氢钠。所述的碱的用量可为有机合成领域此类反应常规的用量,其与通式i-10所示的化合物的摩尔比优选1:1-5:1,更优选2:1-4:1。所述的钯催化剂可为有机合成领域此类反应常规的钯催化剂,优选pd(pph3)2cl2和/或pd(pph3)4。所述的钯催化剂的用量可为有机合成领域此类反应常规的用量,其与通式i-10所示的化合物的摩尔比优选0.01:1-5:1,更优选0.05:1-1.65:1;最优选0.2:1-0.5:1。通式i-10所示的化合物和通式i-11所示的化合物的用量关系可不作具体限定,只要不影响反应进行即可,二者摩尔比优选0.5:1-1:1.5,例如1:1.2。所述的溶剂可为有机合成领域此类反应常规的溶剂,优选水和/或有机溶剂。当所述的溶剂为水和有机溶剂的混合溶剂时,水和有机溶剂的用量可不作具体限定,只要不影响反应进行即可,水和有机溶剂的体积比优选1:3-1:5。所述的溶剂优选除氧处理。所述的有机溶剂优选c1-c4醇类溶剂、芳烃类溶剂和醚类溶剂中的一种或多种。所述的c1-c4醇类溶剂优选乙醇。所述的芳烃类溶剂优选甲苯。所述的醚类溶剂优选1,4-二氧六环。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-10所示的化合物的体积质量比优选1ml/g-50ml/g,优选5ml/g-35ml/g。所述的偶联反应的温度可为有机合成领域此类反应常规的温度,优选常压下溶剂回流温度,例如40-110℃,更优选60-100℃。所述的偶联反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-10所示的化合物消失时作为反应的终点。所述的偶联反应的时间优选30分钟-10小时,更优选2-9小时(例如5-6小时)。
所述的通式i所示的联薁二酰亚胺衍生物的制备方法,其还可进一步包含下列步骤:溶剂中,在亚硝酸类化合物的作用下,将通式i-12所示的化合物和氯化试剂进行如下所示的氯化反应,制得所述的通式i-8所示的化合物;
rax、rbx和rcx的定义均同前所述。
所述的通式i-8所示的化合物的制备方法中,所述的氯化反应的条件可为有机合成领域此类反应常规的条件。其中,所述的亚硝酸类化合物可为有机合成领域此类反应常规的亚硝酸类化合物,优选亚硝酸异戊酯。所述的亚硝酸类化合物的用量可不作具体限定,为有机合成领域此类反应常规用量,其与通式i-12所示的化合物的摩尔比优选1:1-10:1,更优选2:1-5:1。所述的氯化试剂可为有机合成领域此类反应常规的氯化试剂,优选三甲基氯硅烷或氯化氢气体。所述的氯化试剂的用量可不作具体限定,为有机合成领域此类反应常规用量,其与通式i-12所示的化合物的摩尔比优选1:1-10:1,更优选2.5:1-5:1。所述的溶剂可为有机合成领域此类反应常规的溶剂,优选卤代烃类溶剂。所述的卤代烃类溶剂优选氯仿。所述的溶剂的用量可不作具体限定,只要不影响反应进行即可,所述的溶剂与通式i-12所示的化合物的体积质量比优选1ml/g-100ml/g,优选3ml/g-50ml/g(例如3.33ml/g-40.5ml/g)。所述的氯化反应的温度可为有机合成领域此类反应常规的温度,优选室温,例如10-40℃。所述的氯化反应的进程可采用有机合成领域常规的检测方法(例如tlc、gc、1hnmr或gc等)进行监测,一般以通式i-12所示的化合物消失时作为反应的终点。所述的氯化反应的时间优选2-24小时,更优选10-12小时。
本发明中,上述各反应结束后,可采用有机合成领域常规的后处理方法进行后处理。例如水洗、萃取、柱层析等操作。
本发明还提供了一种如式i-1、i-2、i-4或i-5所示的化合物:
其中,x1、x2、r1、r2、ra、rb、rc和rd的定义均同前所述,其中,通式i-2、i-4和i-5所示的化合物中,x1、x2、ra、rb、rc和rd不同时为氢。
本发明还提供了一种所述的通式i所示的化合物在制备有机薄膜场效应晶体管、有机太阳能器件、有机颜料或有机染料中的应用。
所述的应用中,所述的通式i所示的化合物在有机薄膜场效应晶体管中优选作为半导体活性层。所述的通式i所示的化合物在有机太阳能器件中优选作为活性层受体材料。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明中,室温是指10-40℃。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:本发明的联薁二酰亚胺衍生物具有不同的能级、大π共轭体系和柔性促溶的烷基链,可以用溶液加工的方法低成本制备有机电子器件(如otft、opv等);同时本发明的联薁二酰亚胺衍生物制备方法简单,原料易于合成,成本低,得到的目标化合物的纯度高。
附图说明
图1为化合物1和3的紫外-可见吸收光谱图。
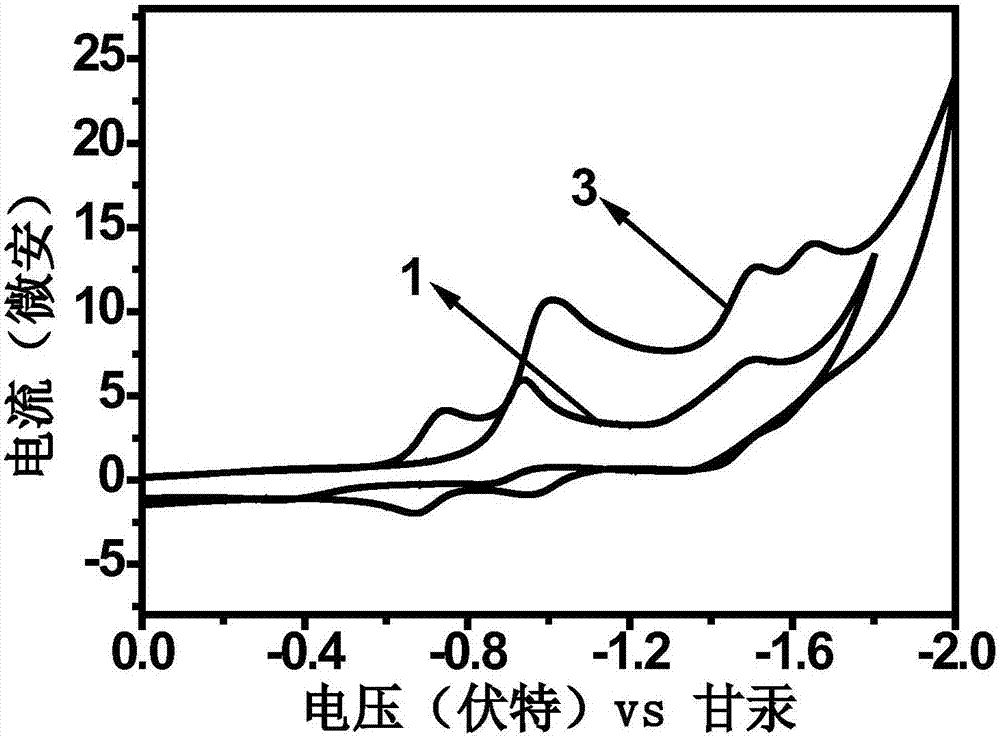
图2为化合物1和3的循环伏安曲线图。
图3为以化合物3作为有机半导体层的otft器件的结构示意图。
图4为化合物3的otft器件的转移曲线图。
图5为以化合物1作为有机半导体层的太阳能电池器件的结构示意图。
图6为化合物1的opv器件的j-v曲线。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
下述实施例中,室温是指10-40℃。过夜是指10-12小时。回流是指常压下溶剂回流温度。
实施例1n,n’-二甲基-2,2’-联薁-1,1’,3,3’-四羧酸二酰亚胺(1)的合成
1.1、2,2’-联薁-1,1’,3,3’-四羧酸乙酯s2
在手套箱中称取化合物s1(921mg,3.0mmol)和ni(cod)2(454mg,1.65mmol)到125ml反应管中。在氮气保护下加入10ml重蒸的dmf。然后避光条件下升温至50℃搅拌反应6h。反应结束后加入少量水,二氯甲烷萃取三次,合并有机相,旋干后柱层析(pe:ea=10:1)分离得红色固体572mg。产率71%。
核磁共振氢谱:1hnmr(400mhz,cdcl3),δ(ppm):9.81(d,j=10.1hz,4h),7.92(t,j=9.7hz,2h),7.74(dd,j=10.1hz,9.7hz,4h),3.89(q,j=7.0hz,8h),0.52(t,j=7.0hz,12h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):165.17,155.33,143.26,139.57,138.20,130.35,116.11,59.29,13.25.低分辨质谱:ms(maldi)m/z:542.0(m)+.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c32h31o8543.2013;实测值:543.2000.
s3的制备:
化合物s2(542mg,1.0mmol)和koh(1.12g,20mmol)溶于20ml乙醇和14ml水中。80℃回流4h后冷却至室温。hcl(2m)中和至有红色固体析出,过滤,固体用水洗,干燥得红色固体386mg。产率90%。
核磁氢谱:1hnmr(300mhz,dmso-d6)δ(ppm):11.86(s,4h),9.71(d,j=10.3hz,4h),8.08(t,j=9.9hz,2h),7.86(dd,j=10.3hz,9.9hz4h).核磁碳谱:13cnmr(100mhz,dmso-d6)δ165.98,155.58,142.48,139.91,137.60,130.12,116.47.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c24h14o8na453.0581;实测值:453.0577.
s4的制备:
化合物s3(430mg,1.0mmol)溶于5ml乙酸酐中。回流4h后冷却至室温。过滤,水洗,干燥得红色固体374mg。产率95%。
元素分析:分子式:c24h10o6;计算值:c,73.10;h,2.56.实测值:c,73.00;h,2.60.
1.2、1’,3’-二(辛基氨基甲酰基)-2,2’-联薁-1,3-二羧酸s5
化合物s4(394mg,1.0mmol)溶于5ml二氯甲烷中,加入正辛胺(387mg,3mmol)到上述溶液中。回流反应4h后冷却至室温。旋干二氯甲烷,柱层析(dcm:etoh=100:1)分离得紫色固体502mg。产率77%。
核磁共振氢谱:1hnmr(400mhz,dmso-d6),δ(ppm):9.55(d,j=10.0hz,2h),9.02(d,j=9.8hz,2h),8.20(t,j=9.8hz,1h),7.95(t,j=10.0hz,1h),7.95(dd,j=10.0hz,10.0hz,2h),7.60(dd,j=10.0hz,9.8hz,2h),6.89(t,j=5.6hz,2h),2.99(m,j=4h).1.20-1.13(m,4h),1.06-0.98(m,4h),0.86-0.59(m,22h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):166.56,166.44,146.54,143.72,142.62,141.16,140.06,137.57,130.70,126.57,120.29,117.76,39.72,31.74,28.98,28.74,28.65,26.36,22.54,14.12.低分辨质谱:ms(maldi)m/z:675.3(m+na)+.高分辨质谱:hrms(maldi-ft)(m/z):(m+h)+计算值:c40h49o6n2653.3585;实测值:543.2000.
1.3、实例化合物1的合成
化合物s5(90mg,0.14mmol)溶于5ml氯化亚砜中,回流反应3h后冷却至室温。旋干氯化亚砜,柱层析(pe:ea=10:1)分离得绿色固体66mg。产率78%。
核磁共振氢谱:1hnmr(400mhz,cdcl3)δ(ppm):9.83(d,j=10.6hz,4h),8.04(t,j=9.3hz,2h),7.82(dd,j=10.6hz,9.3hz,4h),4.40(t,j=7.4hz,4h),1.86(m,4h),1.47–1.26(m,20h),0.86(t,j=6.6hz,6h).核磁共振碳谱:13cnmr(100mhz,cdcl3)δ(ppm):165.91,143.87,142.16,140.71,136.73,131.28,118.57,46.55,31.79,29.43,29.32,29.01,27.38,22.66,14.10.元素分析:分子式:c40h44o4n2;计算值:c,77.89;h,7.19;n,4.54.实测值:c,77.74;h,7.24;n,4.55.
实施例2n,n’-二(2-辛基癸烷基)-6,6’-二(噻吩-2-基)-2,2’-联薁-1,1’,3,3’-四羧酸二酰亚胺(2)的合成
2.1、2-氨基-6-(噻吩2-基)-1,3–薁二羧酸乙酯s7
称取化合物s6(367mg,1.0mmol)、2-噻吩硼酸(200mg,1.5mmo)、乙酸钾(200mg,2mmol)和pd(pph3)2cl2(454mg,1.65mmol)到100ml封管中。在氮气保护下加入10ml除氧的二氧六环和水(5:1)混合溶剂。然后升温至100℃,封管下回流反应6h。反应结束后加入少量水,二氯甲烷萃取三次,合并有机相,旋干后柱层析(pe:ea=10:1)分离得橙红色固体336mg。产率91%。
核磁共振氢谱:1hnmr(300mhz,cdcl3),δ(ppm):9.08(d,j=10.7hz,2h),7.89(d,j=10.7hz,2h),7.79(s,2h),7.46(d,j=3.2hz,1h),7.39(d,j=5.2hz,1h),7.14(dd,j=3.2hz,j=5.2hz,1h),4.47(q,j=7.0hz,4h),1.49(t,j=7.0hz,6h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):166.40,162.25,146.66,144.37,138.45,130.46,130.27,128.62,126.94,125.29,100.22,59.83,14.67.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c20h19o4ns370.1108;实测值:370.1104.
2.2、2-氯-6-(噻吩2-基)-1,3–薁二羧酸乙酯s8
化合物s7(740mg,2mmol)、三甲基氯硅烷(1.08g,5mmol)、亚硝酸异戊酯(1.17g,5mmol)加入到装完30ml氯仿的100ml反应瓶中,室温搅拌反应12h,旋干体系中的氯仿,柱层析(pe:dcm=1:2)分离得紫红色固体590mg。产率76%。
核磁共振氢谱:1hnmr(300mhz,cdcl3),δ(ppm):9.38(d,j=11.2hz,2h),7.99(d,j=11.2hz,2h),7.60(d,j=2.7hz,1h),7.52(d,j=5.0hz,1h),7.18(dd,j=2.7hz,j=5.0hz,1h),4.48(q,j=7.1hz,4h),1.48(t,j=7.1hz,6h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):164.18,145.82,145.59,142.56,139.93,136.99,129.60,129.19,128.49,127.74,115.62,60.62,14.45.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c20h18o4cls389.0609;实测值:389.0607.
2.3、6,6’-二(噻吩-2-基)-2,2’-联薁-1,1’,3,3’-四羧酸乙酯s9
在手套箱中称取化合物s8(388mg,1.0mmol)和ni(cod)2(152mg,0.55mmol)到100ml反应管中。在氮气保护下加入8ml重蒸的dmf。然后避光条件下升温至50℃搅拌反应6h。反应结束后加入少量水,二氯甲烷萃取三次,合并有机相,旋干后柱层析(pe:dcm=1:2)分离得红色固体328mg。产率93%。
核磁共振氢谱:1hnmr(400mhz,cdcl3),δ(ppm):9.74(d,j=11.3hz,4h),8.08(d,j=11.3hz,4h),7.66(d,j=2.9hz,2h),7.51(d,j=5.1hz,2h),7.19(dd,j=2.9hz,j=5.1hz,2h),3.94(q,j=7.1hz,8h),0.61(t,j=7.1hz,12h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):165.19,154.79,146.30,145.19,141.76,137.33,129.09,129.05,128.11,127.35,116.75,59.37,13.41.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c40h35o8s2707.1768;实测值:707.1764.
2.4、6,6’-二(噻吩-2-基)-2,2’-联薁-1,1’,3,3’-四羧酸s10
取化合物s9(500mg,0.7mmol)和氢氧化钾(785mg,14mmol)加入到100ml反应瓶中,加入12ml乙醇、1.5ml水和12ml四氢呋喃,加热到80℃回流反应10小时。冷却至室温,用2m盐酸中和至中性,析出固体,过滤、水洗、真空干燥得红色产物404mg。产率95%。
高分辨质谱:hrms(maldi-ft)(m/z):(m+h)+计算值:c32h19o8s2595.0516;实测值:595.0514.
2.5、6,6’-二(噻吩-2-基)-2,2’-联薁-1,1’,3,3’-四羧酸二酐s11
取化合物s10(300mg,0.5mmol)加入到50ml反应瓶中,加入12ml乙酸酐,加热到140℃回流反应4小时。冷却至室温,过滤、水洗、真空干燥得红色产物254mg。产率91%。
元素分析:分子式:c32h14o6s2;计算值:c,68.81;h,2.53.实测值:c,68.50;h,2.84.
2.6、实例化合物2的合成
化合物s11(280mg,0.5mmol)加入到50ml反应瓶中,加入15ml二氯甲烷和2-己基癸胺(482mg,2mmol),加热到80℃回流反应5小时。冷却至室温,旋干二氯甲烷,快速柱层析分离(dcm:meoh=20:1)得绿色产物,未经进一步纯化直接加入4ml二氯亚砜,80℃回流2小时。旋干二氯亚砜后柱层析(pe:dcm=1:2)分离得红色产物265mg。产率53%。
核磁共振氢谱:1hnmr(300mhz,cdcl3),δ(ppm):δ9.61(d,j=11.2hz,4h),8.06(d,j=11.2hz,4h),7.65(d,j=3.3hz,2h),7.53(d,j=4.8hz,2h),7.17(dd,j=3.3hz,j=4.8hz,2h),4.41(d,j=6.8hz,4h),2.07(m,2h),1.50–1.02(m,48h),0.81(m,12h).核磁共振碳谱:13cnmr(100mhz,cdcl3),δ(ppm):166.09,147.24,145.67,142.21,139.30,135.93,130.16,129.29,128.38,128.14,118.99,50.15,37.26,31.89,31.84,30.22,29.87,29.62,29.41,26.46,22.71,22.68,14.09.高分辨质谱:hrms(maldi-ft)(m/z):(m+h)+计算值:c64h81o8n2s21005.5632;实测值:1005.5633.元素分析:分子式:c64h80o8n2s2;计算值:c,76.45;h,8.02;n,2.79.实测值:c,76.27;h,8.22;n,2.58.
实施例3n,n’-二(2-辛基癸烷基)-2,6’:2’,2”:6”,2”-四薁-1,1’,3,3’-四羧酸二酰亚胺(3)的合成
3.1、2-氯-6-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)-1,3-薁二羧酸乙酯s14
化合物s13(1.24g,3mmol)、三甲基氯硅烷(3.24g,15mmol)、亚硝酸异戊酯(3.51g,15mmol)加入到装完80ml氯仿的250ml反应瓶中,室温搅拌反应12h,旋干体系中的氯仿,柱层析(pe:dcm=1:2)分离得紫色固体1.12g。产率86%。
核磁共振氢谱:1hnmr(400mhz,cdcl3)δ(ppm):9.44(d,j=10.2hz,2h),8.21(d,j=10.2hz,2h),4.47(d,j=7.1hz,4h),1.46(t,j=7.1hz,6h).核磁共振碳谱:13cnmr(100mhz,cdcl3)δ(ppm):164.18,144.84,142.65,136.83,115.04,85.15,60.64,24.90,14.41.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c22h27o6bcl432.1620;实测值:432.1616.
3.2、2-氯-2,6’-联薁-1,3–二羧酸乙酯s15
称取化合物s14(1.56g,3.6mmol)、2-溴代薁(414mg,2.0mmol)、碳酸氢钠(1.09g,13mmol)和pd(pph3)2cl2(140mg,0.2mmol)到100ml反应瓶中。在氮气保护下加入4ml除氧的水、4ml除氧的乙醇和8ml除氧的甲苯。然后升温至60℃反应2h。反应结束后加入少量水,二氯甲烷萃取三次,合并有机相,旋干后柱层析(pe:dcm=1:2)分离得绿色固体510mg。产率59%。
核磁共振氢谱:1hnmr(300mhz,cdcl3),δ(ppm):9.38(d,j=11.2hz,2h),7.99(d,j=11.2hz,2h),7.60(d,j=2.7hz,1h),7.52(d,j=5.0hz,1h),7.18(dd,j=2.7hz,j=5.0hz,1h),4.48(q,j=7.1hz,4h),1.48(t,j=7.1hz,6h).核磁共振氢谱:13cnmr(100mhz,cdcl3),δ(ppm):164.18,145.82,145.59,142.56,139.93,136.99,129.60,129.19,128.49,127.74,115.62,60.62,14.45.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c20h18o4cls389.0609;实测值:389.0607.
3.3、2,6’:2’,2”:6”,2”-四薁-1,1’,3,3’-四羧酸乙酯s16
在手套箱中称取化合物s15(1.68g,3.7mmol)和ni(cod)2(0.76g,2.8mmol)到100ml反应管中。在氮气保护下加入12ml无水的dmf。然后避光条件下升温至50℃搅拌反应6h。反应结束后加入少量水,二氯甲烷萃取三次,合并有机相,旋干后柱层析(pe:dcm=1:4)分离得绿色固体1.49g。产率97%。
核磁共振氢谱:1hnmr(300mhz,cdcl3)δ(ppm):9.86(d,j=10.8hz,4h),8.43(d,j=10.8hz,4h),8.41(d,j=9.6hz,4h),7.86(s,4h),7.62(t,j=10.0hz,2h),7.23(dd,j=10.0hz,9.6hz,4h),3.97(q,j=7.0hz,8h),0.64(t,j=7.0hz,12h).核磁共振碳谱:13cnmr(100mhz,cdcl3)δ(ppm):165.32,155.37,151.07,147.79,142.33,141.48,138.43,138.02,137.23,130.42,124.50,116.49,116.41,59.36,13.42.高分辨质谱:hrms(dart-ft)(m/z):(m+h)+计算值:c52h43o8795.2952;实测值:795.2945.
3.4、2,6’:2’,2”:6”,2”-四薁-1,1’,3,3’-四羧酸s17
取化合物s16(794mg,1.0mmol)和氢氧化钾(1.12g,20mmol)加入到100ml反应瓶中,加入16ml乙醇、2ml水和16ml四氢呋喃,加热到80℃回流反应24小时。冷却至室温,用2m盐酸中和至中性,析出固体,过滤、水洗、真空干燥得棕色产物636mg。产率83%。
高分辨质谱:hrms(esinegative)(m/z):(m–h)–计算值:c44h25o8681.1555;实测值:681.1544.
3.5、2,6’:2’,2”:6”,2”-四薁-1,1’,3,3’-四羧酸二酐s18
取化合物s17(205mg,0.3mmol)加入到25ml反应瓶中,加入5ml乙酸酐,加热到140℃回流反应2小时。冷却至室温,过滤、水洗、真空干燥得棕色产物169mg。产率87%。
质谱:ms(maldi)m/z:647.2(m+h)+.
3.6、实例化合物3的合成
化合物s18(65mg,0.1mmol)加入到50ml反应瓶中,加入15ml二氯甲烷和2-己基癸胺(73mg,0.3mmol),加热到80℃回流反应4小时。冷却至室温,旋干二氯甲烷,快速柱层析分离(dcm:meoh=20:1)得绿色产物,未经进一步纯化直接加入10ml乙酸酐和乙酸钠(82mg,1.0mmol),140℃回流4小时。反应结束后,二氯甲烷萃取,柱层析(pe:dcm=2:1)分离得红色产物46mg。产率42%。
核磁共振氢谱:1hnmr(400mhz,cdcl3)δ(ppm):9.63(d,j=11.2hz,4h),8.30(d,j=11.2hz,4h),8.27(d,j=9.3hz,4h),7.68(s,4h),7.50(t,j=9.9hz,2h),7.12(dd,j=9.9hz,9.3hz,4h),4.45(d,j=7.5hz,4h),2.10(s,2h),1.29(m,48h),0.79(t,j=6.7hz,12h).核磁共振碳谱:13cnmr(100mhz,cdcl3)δ165.65,149.40,149.31,142.18,140.87,138.57,138.32,137.92,135.76,130.25,124.03,118.19,116.01,49.69,36.85,31.39,29.78,29.42,29.17,28.95,26.01,22.23,22.19,13.61.高分辨质谱:hrms(maldi-ft)(m/z):(m+h)+:计算值:c76h89o4n21093.6817;实测值:1093.6812.元素分析:分子式:c76h88o4n2;计算值:c,83.47;h,8.11;n,2.56.实测值:c,83.61;h,8.03;n,2.39.
实施例4n,n’-二(2-辛基癸烷基)-6,6’-二(4-溴噻吩-2-基)-2,2’-联薁-1,1’,3,3’-四羧酸二酰亚胺(4)的合成
称取化合物3(56mg,0.05mmol)加入到50ml反应瓶中,加入10ml二氯甲烷,加入液溴(22.4mg,0.15mmol)。室温下搅拌反应12小时。反应结束后二氯甲烷萃取,柱层析(pe:dcm=2:1)分离的产物58mg。产率90%。
高分辨质谱:hrms(maldi-ft)(m/z):(m+h)+:计算值:c64h79o4n2br2s21161.3843;实测值:1161.3822.
效果实施例1化合物1和3的紫外吸收光谱和电化学性质
紫外吸收光谱在u-3900光谱仪上进行,样品溶液二氯甲烷中(摩尔浓度为1×10-6m),扫描范围为800-200nm,化合物的光学带隙由以下公式计算得到:
egapopt=1240nm/λonset(1)
循环伏安法测试在计算机控制的chi610d电化学分析仪上进行,采用传统的三电极测试体系,铂电极为工作电极,饱和甘汞电极(sce)作为参比电极,铂丝作为对电极,样品溶于新蒸的二氯甲烷(摩尔浓度为1×10-3m),bu4npf6(0.1m)作为支持电解质,扫描速度为50mv/s,以饱和甘汞为参比,饱和甘汞能级相对于真空能级为-4.44ev,材料的lumo能级可以由以下能级的公式计算得到:
elumo=-(e1/2red1+4.44)ev(2)
图1为化合物1和3的紫外-可见吸收光谱图,其起始吸收峰分别在667nm和734nm左右,由公式(1)计算得到其光学带隙分别为1.86ev和1.69ev。图2为化合物1和3的循环伏安曲线图,由循环伏安曲线及公式(2)可以计算化合物1和3的lumo能级分别为-3.53ev和-3.74ev。
效果实施例2化合物3作为半导体活性层制备有机薄膜场效应晶体管
图3为以化合物3作为有机半导体层的otft器件的结构示意图。otft器件的制备方法为:将5-15mg的化合物3溶于1ml氯仿中,在ots(辛基三氯硅烷)修饰的sio2/si基底上(高掺杂的硅衬底作为栅极,热氧化二氧化硅绝缘层的厚度为450nm,电容为10nfcm-2)甩上一层约20-80nm厚度的有机半导体薄膜,在有机薄膜的上面利用掩模板沉积金源漏电极,从而制得上电极结构的otft器件,器件的半导体沟道长度为50μm,沟道宽度为3mm。otft的电性能用keithley4200半导体测试仪在氮气中室温下测到。图4为化合物3的otft器件的转移曲线图,其电子迁移率为0.015cm2v-1s-1,开关比为104-105。
效果实施例3化合物1作为半导体活性层制备有机太阳能电池器件
图5为以化合物1作为有机半导体层的太阳能电池器件的结构示意图。太阳能电池器件的制备方法为:以p3ht(聚三己基噻吩)为给体材料,所合成的化合物1为受体材料,制备有机太阳能电池。在洗干净的ito导电玻璃基片上,旋涂30nm厚度的pedot:pss(聚3,4-乙撑二氧噻吩/聚苯乙烯磺酸盐)导电膜。干燥处理后,将ito(氧化铟锡)导电玻璃基片转移至手套箱中,旋涂100nm厚度的活性层膜。活性层材料中给受体的共混比例为1:1(w:w),选用氯苯或邻二氯苯作溶剂。将旋涂好活性层的ito玻璃基片转移至120℃的热台上退火处理10分钟,以使活性层达到较好的微相形貌。再将热处理好的旋涂了活性层的ito玻璃基片转移至蒸镀仓中,先蒸镀一层20nm厚度的ca,再蒸镀一层100nm厚度的al,制备出结构为ito/pedot:pss/p3ht:xx/ca/al的有机太阳能电池器件,电池面积为7mm2。用orielso13a太阳光模拟器对所制备的电池器件进行i-v特性测试。图6为化合物1的opv器件的j-v曲线,其太阳能转化效率为0.27%。
- 还没有人留言评论。精彩留言会获得点赞!
- 低介电常数树脂基材用处理铜箔及使用该处理铜箔的覆铜层压板以及印刷配线板的制造方法与工艺
- 含有色酮结构的吡唑类N‐对溴苯基马来酰亚胺衍生物及其制备方法与应用与制造工艺
- 一种双吲哚马来酰亚胺生物碱HD‑ZWM288在制备抗肿瘤微管抑制剂药物的应用的制造方法与工艺
- 马来酰亚胺膜的制造方法与工艺
- 多孔性酰亚胺系树脂膜制造系统、隔膜、及多孔性酰亚胺系树脂膜制造方法与制造工艺
- 聚苯并噁嗪前体及其制备方法与制造工艺
- 含酰亚胺结构的环三磷腈无卤阻燃剂、制备方法及用途与制造工艺
- 开关型硫醇荧光标记试剂及其合成方法和应用与制造工艺
- 一种耐高温表面自润滑的聚酰胺酰亚胺绝缘漆的制造方法与工艺
- 一种耐高温表面自润滑的聚酰胺酰亚胺绝缘漆的制造方法与工艺