一种盐酸左西替利嗪片的有关物质及其分析检测方法与流程
本发明属于药物制剂分析领域,具体涉及一种盐酸左西替利嗪片的有关物质及其分析检测方法。
背景技术:
盐酸左西替利嗪片为口服选择性组胺H1受体拮抗剂,是新一代高效非镇静抗组胺剂。盐酸左西替利嗪是盐酸西替利嗪的旋光体,适用于季节性过敏性鼻炎、长年过敏性鼻炎和荨麻疹的治疗,也用于减轻感冒时的过敏症状。在临床上比盐酸西替利嗪具有更高的药效、更好的安全性。盐酸西替利嗪R-异构体对H1-受体的亲和力是盐酸西替利嗪的2倍,为高选择性外周H1-受体拮抗剂。盐酸左西替利嗪于2001年由比利时UCB公司首先于德国上市,随后又在英国上市,2005年在法国上市。
盐酸左西替利嗪片的活性成分为盐酸左西替利嗪,其化学结构式如下:
技术实现要素:
本发明的目的在于提供一种盐酸左西替利嗪片的有关物质及其分析检测方法。
本发明的上述目的是通过下面的技术方案得以实现的:
一种盐酸左西替利嗪片中的有关物质,化学结构式如下:
上述有关物质的制备方法,包括如下步骤:
步骤S1,高温破坏:将盐酸左西替利嗪原料药置于120~140℃条件下破坏24~36小时;
步骤S2,富集:将上述高温破坏样品溶于甲醇体积百分浓度为75%~85%的甲醇水溶液中,搅拌,抽滤,收集滤液,减压浓缩得浓缩物;
步骤S3,粗品制备:将上述浓缩物用正相硅胶柱色谱分离,依次用体积比为20:1、10:1和5:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集5:1洗脱部分,浓缩得粗品;
步骤S4,精制:将上述粗品用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为45%~55%的乙腈水溶液,等度洗脱,收集第6~10个柱体积洗脱液,浓缩,冷冻干燥即得所述有关物质。
进一步地,所述的制备方法包括如下步骤:
步骤S1,高温破坏:将盐酸左西替利嗪原料药置于130℃条件下破坏30小时;
步骤S2,富集:将上述高温破坏样品溶于甲醇体积百分浓度为80%的甲醇水溶液中,搅拌,抽滤,收集滤液,减压浓缩得浓缩物;
步骤S3,粗品制备:将上述浓缩物用正相硅胶柱色谱分离,依次用体积比为20:1、10:1和5:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集5:1洗脱部分,浓缩得粗品;
步骤S4,精制:将上述粗品用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为50%的乙腈水溶液,等度洗脱,收集第7~9个柱体积洗脱液,浓缩,冷冻干燥即得所述有关物质。
上述有关物质的分析方法,包括如下参数:
色谱柱:Hypersil BDS C18(4.6mm×250mm,5μm);
流动相:A为乙腈,B为含0.02mol/L磷酸二氢铵的水溶液(磷酸调节pH值至3.5);
梯度洗脱程序:0~5min,A 28%;5~30min,A 28%→75%;30~35min,A 75%;35~40min,A 75%→28%;40~45min,A 28%;
流动相流速:1.0mL·min-1;
检测波长:230nm;
柱温:35℃;
进样量:10μL。
上述有关物质在盐酸左西替利嗪片有关物质检查中作为对照品的用途。
本发明的优点:
发明人在盐酸左西替利嗪片有关物质研究过程中,通过调整色谱条件,检测到一个与盐酸左西替利嗪色谱保留行为非常接近的色谱峰,并制备出了该色谱峰对应的化学标准品,经鉴定为本发明提供的有关物质。该有关物质为首次报道,在盐酸左西替利嗪原料药和片剂中均可检测到该成分,且含量较高(大于万分之五),有必要对其进行分析控制。
附图说明
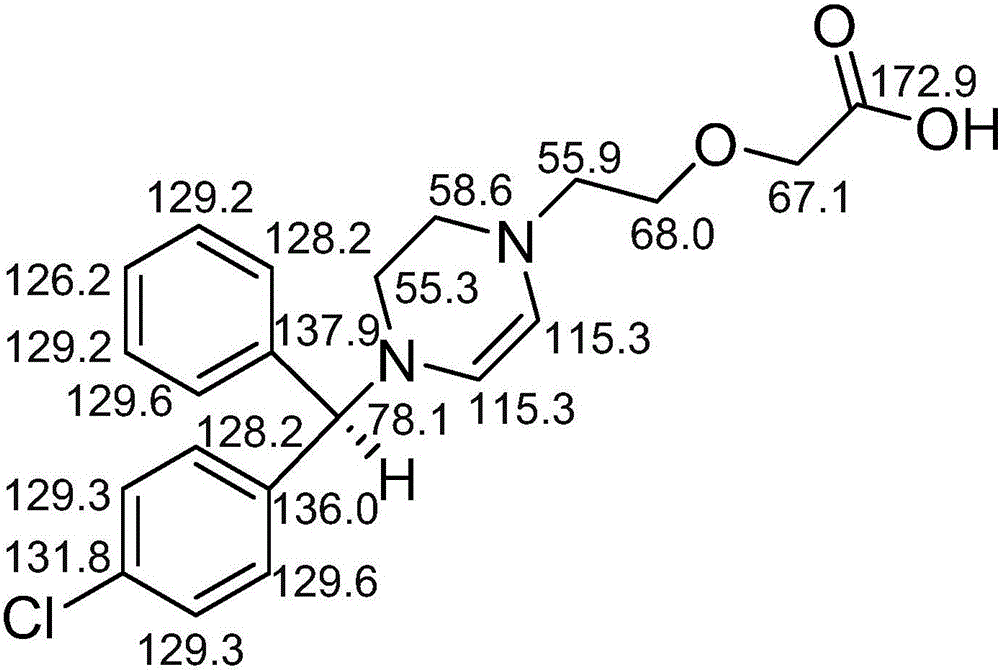
图1为本发明有关物质氢谱数据归属图;
图2为本发明有关物质碳谱数据归属图。
具体实施方式
下面结合实施例进一步说明本发明的实质性内容,但并不以此限定本发明保护范围。尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
实施例1:有关物质的制备和结构确证
取10g盐酸左西替利嗪原料均匀摊在培养皿中,置于烘箱中,130℃破坏30小时。然后,将高温破坏样品溶于甲醇体积百分浓度为80%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物7.3g;然后将浓缩物用10ml甲醇溶解,用20g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为20:1、10:1和5:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集5:1洗脱部分(每个比例洗脱10个柱体积),减压浓缩得粗品4.5g;然后将粗品用乙腈体积百分浓度为50%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为50%的乙腈-水,等度洗脱,收集第7~9个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末2.3g。HPLC归一化纯度大于98%。
结构确证:
HR-ESIMS显示[M+Na]+为m/z 409.1312,结合核磁特征可得分子式为C21H23ClN2O3。氢谱数据及归属见图1(ppm,methanol-d4,500MHz);碳谱数据及归属见图2(ppm,methanol-d4,150MHz)。综合质谱和核磁数据,结合左西替利嗪的结构确证数据可确证该物质的化学结构式如下所示:
实施例2:有关物质的制备
取10g盐酸左西替利嗪原料均匀摊在培养皿中,置于烘箱中,120℃破坏36小时。然后,将高温破坏样品溶于甲醇体积百分浓度为75%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物7.2g;然后将浓缩物用10ml甲醇溶解,用20g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为20:1、10:1和5:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集5:1洗脱部分(每个比例洗脱10个柱体积),减压浓缩得粗品4.3g;然后将粗品用乙腈体积百分浓度为45%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为45%的乙腈-水,等度洗脱,收集第8~10个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末2.2g。HPLC归一化纯度大于98%。
实施例3:有关物质的制备
取10g盐酸左西替利嗪原料均匀摊在培养皿中,置于烘箱中,140℃破坏24小时。然后,将高温破坏样品溶于甲醇体积百分浓度为85%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物7.4g;然后将浓缩物用10ml甲醇溶解,用20g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为20:1、10:1和5:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集5:1洗脱部分(每个比例洗脱10个柱体积),减压浓缩得粗品4.6g;然后将粗品用乙腈体积百分浓度为55%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为55%的乙腈-水,等度洗脱,收集第6~8个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末2.1g。HPLC归一化纯度大于98%。
实施例4:盐酸左西替利嗪片加速试验(40℃条件下30天)样品检测分析
盐酸左西替利嗪片:收集市售不同品牌的盐酸左西替利嗪片(距生产日期3个月内)。
对照品溶液制备:精密称取2mg本发明有关物质于200ml容量瓶中,用50%乙腈溶解定容,作为有关物质储备液;精密量取有关物质储备液1ml于20ml容量瓶中,用液相分析方法中初始比例的流动相稀释定容(约0.5μg/mL)。
供试品溶液制备:取40℃条件下放置30天的盐酸左西替利嗪片研磨成细粉,精密称取细粉适量(约含左西替利嗪12.5mg)置于25ml容量瓶中,添加液相分析方法中初始比例的流动相,超声使左西替利嗪溶解,放冷,稀释至刻度。
对照溶液:精密量取供试品溶液0.5ml于200ml容量瓶中,用液相分析方法中初始比例的流动相稀释至刻度。
液相色谱条件:
高效液相色谱仪:Agilent 1260,二元泵,VWD;
色谱柱:Hypersil BDS C18(4.6mm×250mm,5μm);
流动相:A为乙腈,B为含0.02mol/L磷酸二氢铵的水溶液(磷酸调节pH值至3.5);
梯度洗脱程序:0~5min,A 28%;5~30min,A 28%→75%;30~35min,A 75%;35~40min,A 75%→28%;40~45min,A 28%;
流动相流速:1.0mL·min-1;
检测波长:230nm;
柱温:35℃;
进样量:10μL。
分别精密量取有关物质对照品溶液、供试品溶液和对照溶液10μL注入液相色谱仪分析。结果在不同品牌盐酸左西替利嗪片中均检测到本发明提供的有关物质(保留时间为左西替利嗪保留时间1.1倍),该有关物质的色谱峰与左西替利嗪色谱峰分离度均大于2.0,且峰纯度检查合格。不同品牌盐酸左西替利嗪片中该有关物质的含量为0.05~0.15%,有必要列入盐酸左西替利嗪片的质量研究控制中进行进一步研究(如长期稳定性考察,毒理考察),以提高盐酸左西替利嗪片的质量标准。
上述实施例的作用在于说明本发明的实质性内容,但并不以此限定本发明的保护范围。本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和保护范围。
- 还没有人留言评论。精彩留言会获得点赞!