一种磷酸奥司他韦异构体杂质的合成方法与流程
本发明属于医药化工领域,具体涉及一种磷酸奥司他韦异构体杂质的合成方法。
背景技术:
磷酸奥司他韦其化学名称为(3R,4R,5S)-4-乙酰氨基-5-氨基-3-(1-乙基丙氧基)-1-环己烯羧酸乙酯,被认为是目前最有效、特异性最高的流感治疗药物,可以大大减少并发症(主要是气管与支气管炎、肺炎、咽炎等)的发生和抗生素的使用,因而是目前治疗流感的最常用药物之一,也是公认的抗禽流感、甲型病毒最有效的药物之一,于1999年被美国批准上市,在中国于2004年7月上市。
化合物Ⅰ为磷酸奥司他韦的异构体杂质,EP中命名为杂质G,是磷酸奥司他韦生产过程中的一个常见的杂质。合成该杂质研究其理化性质从而加以控制并选择合适方法有效去除对于提高磷酸奥司他韦质量意义重大。本专利以申请号CN88106883.7(CN1033044A)中的中间体作为起始原料,在手性路易斯酸的作用下与苄胺及其衍生物进行选择性反应,并通过成盐纯化后氢解得到目标化合物。
技术实现要素:
本发明主要涉及一种磷酸奥司他韦异构体杂质的合成方法,有利于提高磷酸奥司他韦原料药的质量控制。
本发明技术方案:
一种磷酸奥司他韦异构体杂质的合成方法,其结构如下:
按照以下路线进行合成:
主要包含以下三个步骤:
1)以化合物为Ⅱ起始原料,在溶剂1中,与化合物Ⅲ在手性路易斯酸催化剂作用下进行反,得到物料1;
2)滤掉所述步骤1)物料1中的催化剂并蒸干溶剂1后,在溶剂2中与有机酸成盐纯化后的固体,加入二氯甲烷-20%液碱体系下解离后得到化合物Ⅳ;
3)化合物Ⅳ在溶剂3中通过10%钯碳,Raney-Ni,5%铂碳催化剂室温下氢解脱去氨基保护基并成磷酸盐,并通过丙酮-庚烷重结晶得到化合物Ⅰ。
优选地,所述化合物Ⅲ、化合物Ⅳ中R1基团为H或CH3或CH2CH3。
优选地,所述步骤1)手性路易斯催化剂为(S)-(-)-BINOL-Ti或(S)-(-)-BINOL-Mg络合物。
进一步优选地,所述(S)-(-)-BINOL-Ti为摩尔比为1:1-1:2的S-BINOL和Ti(i-OPr)4制备的络合物,所述(S)-(-)-BINOL-Mg为摩尔比为1:1-1:2的S-BINOL和MgCl2制备的络合物。
优选地,所述步骤1)溶剂1为甲苯或二甲苯或四氯化碳,所述步骤1)中化合物为Ⅱ和化合物Ⅲ的摩尔比1:0.8-1:1.2,所述溶剂1是化合物为Ⅱ重量的8-12倍,所述手性路易斯酸催化剂为化合物为Ⅱ重量的0.05-0.15倍。
优选地,所述步骤2)溶剂2为甲醇或乙醇或异丙醇或丙酮,有机酸为L-脯氨酸或D-酒石酸或马来酸。
进一步优选地,所述步骤2)物料1与有机酸体积比为1:1-1:1.5,所述成盐纯化后的固体加入二氯甲烷与20%氢氧化钠体积比为2:1的溶液中,于25-30℃解离后得到化合物Ⅳ。
优选地,所述步骤3)中氢解使用的溶剂为甲醇或异丙醇或乙醇。
进一步优选地,所使用的催化剂为10%钯碳,Raney-Ni,5%铂碳,用量分别为化合物Ⅳ重量的2%-3%、3%-5%、1%-2%。
优选地,所述步骤3)重结晶所采用的体系为丙酮-庚烷,丙酮与庚烷比例为1:4-1:10。
本发明的有益效果:
提供一个简单方便的异构体杂质的合成方法,通过对其性质的研究,有利于提高磷酸奥司他韦原料药的质量控制。
附图说明
图1,磷酸奥司他韦异构杂质的1H-NMR图谱
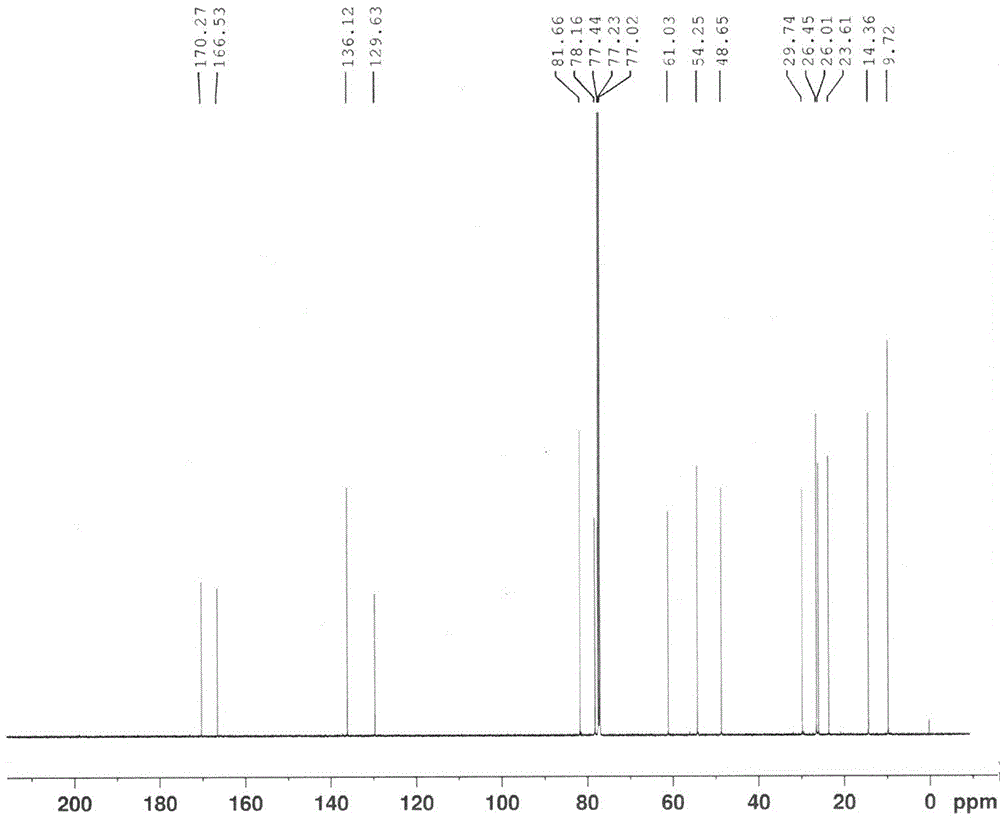
图2,磷酸奥司他韦异构杂质的13C-NMR图谱
具体实施方式
下面结合实施例来进一步说明本发明,但本发明要求保护的范围并不局限于实施例表述的范围。
实施例1
一种磷酸奥司他韦异构体杂质的合成方法,其结构如下:
按照以下路线进行合成:
主要包含以下三个步骤:
1)以化合物为Ⅱ起始原料,在溶剂1中,与化合物Ⅲ在手性路易斯酸催化剂作用下进行反,得到物料1;
2)滤掉所述步骤1)物料1中的催化剂并蒸干溶剂1后,在溶剂2中与有机酸成盐纯化后的固体,加入二氯甲烷-20%液碱体系下解离后得到化合物Ⅳ;
3)化合物Ⅳ在溶剂3中通过10%钯碳,Raney-Ni,5%铂碳催化剂室温下氢解脱去氨基保护基并成磷酸盐,并通过丙酮-庚烷重结晶得到化合物Ⅰ。
所述化合物Ⅲ、化合物Ⅳ中R1基团为H或CH3或CH2CH3。
所述步骤1)手性路易斯催化剂为(S)-(-)-BINOL-Ti或(S)-(-)-BINOL-Mg络合物。
所述(S)-(-)-BINOL-Ti为摩尔比为1:1的S-BINOL和Ti(i-OPr)4制备的络合物,所述(S)-(-)-BINOL-Mg为摩尔比为1:1的S-BINOL和MgCl2制备的络合物。
所述步骤1)溶剂1为甲苯或二甲苯或四氯化碳,所述步骤1)中化合物为Ⅱ和化合物Ⅲ的摩尔比1:0.8,所述溶剂1是化合物为Ⅱ重量的8倍,所述手性路易斯酸催化剂为化合物为Ⅱ重量的0.05倍。
所述步骤2)溶剂2为乙醇,有机酸为D-酒石酸。
所述步骤2)物料1与有机酸体积比为1:1,所述成盐纯化后的固体加入二氯甲烷与20%氢氧化钠体积比为2:1的溶液中,于25℃解离后得到化合物Ⅳ。
所述步骤3)中氢解使用的溶剂为异丙醇。
所使用的催化剂为10%钯碳,Raney-Ni,5%铂碳,用量分别为化合物Ⅳ重量的2%、3%、1%。
所述步骤3)重结晶所采用的体系为丙酮-庚烷,丙酮与庚烷比例为1:4。
实施例2
按照实施例1合成路径,合成方法如下:
1)以化合物为Ⅱ起始原料,在溶剂1中,与化合物Ⅲ在手性路易斯酸催化剂作用下进行反,得到物料1;
2)滤掉所述步骤1)物料1中的催化剂并蒸干溶剂1后,在溶剂2中与有机酸成盐纯化后的固体,加入二氯甲烷-20%液碱体系下解离后得到化合物Ⅳ;
3)化合物Ⅳ在溶剂3中通过10%钯碳,Raney-Ni,5%铂碳催化剂室温下氢解脱去氨基保护基并成磷酸盐,并通过丙酮-庚烷重结晶得到化合物Ⅰ。
所述化合物Ⅲ、化合物Ⅳ中R1基团为H或CH3或CH2CH3。
所述步骤1)手性路易斯催化剂为(S)-(-)-BINOL-Ti或(S)-(-)-BINOL-Mg络合物。
所述(S)-(-)-BINOL-Ti为摩尔比为1:2的S-BINOL和Ti(i-OPr)4制备的络合物,所述(S)-(-)-BINOL-Mg为摩尔比为1:2的S-BINOL和MgCl2制备的络合物。
所述步骤1)溶剂1为甲苯或二甲苯或四氯化碳,所述步骤1)中化合物为Ⅱ和化合物Ⅲ的摩尔比1:1.2,所述溶剂1是化合物为Ⅱ重量的12倍,所述手性路易斯酸催化剂为化合物为Ⅱ重量的0.15倍。
所述步骤2)溶剂2为丙酮,有机酸为马来酸。
所述步骤2)物料1与有机酸体积比为1:1.5,所述成盐纯化后的固体加入二氯甲烷与20%氢氧化钠体积比为2:1的溶液中,于30℃解离后得到化合物Ⅳ。
所述步骤3)中氢解使用的溶剂为乙醇。
所使用的催化剂为10%钯碳,Raney-Ni,5%铂碳,用量分别为化合物Ⅳ重量的3%、5%、2%。
所述步骤3)重结晶所采用的体系为丙酮-庚烷,丙酮与庚烷比例为1:10。
实施例3
按照实施例1合成路径,合成方法如下:
1)以化合物为Ⅱ起始原料,在溶剂1中,与化合物Ⅲ在手性路易斯酸催化剂作用下进行反,得到物料1;
2)滤掉所述步骤1)物料1中的催化剂并蒸干溶剂1后,在溶剂2中与有机酸成盐纯化后的固体,加入二氯甲烷-20%液碱体系下解离后得到化合物Ⅳ;
3)化合物Ⅳ在溶剂3中通过10%钯碳,Raney-Ni,5%铂碳催化剂室温下氢解脱去氨基保护基并成磷酸盐,并通过丙酮-庚烷重结晶得到化合物Ⅰ。
所述化合物Ⅲ、化合物Ⅳ中R1基团为H或CH3或CH2CH3。
所述步骤1)手性路易斯催化剂为(S)-(-)-BINOL-Ti或(S)-(-)-BINOL-Mg络合物。
所述(S)-(-)-BINOL-Ti为摩尔比为1:1.5的S-BINOL和Ti(i-OPr)4制备的络合物,所述(S)-(-)-BINOL-Mg为摩尔比为1:1.5的S-BINOL和MgCl2制备的络合物。
所述步骤1)溶剂1为甲苯或二甲苯或四氯化碳,所述步骤1)中化合物为Ⅱ和化合物Ⅲ的摩尔比1:1,所述溶剂1是化合物为Ⅱ重量的10倍,所述手性路易斯酸催化剂为化合物为Ⅱ重量的0.1倍。
所述步骤2)溶剂2为异丙醇,有机酸为D-酒石酸。
所述步骤2)物料1与有机酸体积比为1:1.4,所述成盐纯化后的固体加入二氯甲烷与20%氢氧化钠体积比为2:1的溶液中,于28℃解离后得到化合物Ⅳ。
所述步骤3)中氢解使用的溶剂为甲醇或异丙醇或乙醇。
所使用的催化剂为10%钯碳,Raney-Ni,5%铂碳,用量分别为化合物Ⅳ重量的2.5%、4%、1.5%。
所述步骤3)重结晶所采用的体系为丙酮-庚烷,丙酮与庚烷比例为1:6。
实施例4
按照实施例1合成路径,合成方法如下:
1)以化合物为Ⅱ起始原料,在溶剂1中,与化合物Ⅲ在手性路易斯酸催化剂作用下进行反,得到物料1;
2)滤掉所述步骤1)物料1中的催化剂并蒸干溶剂1后,在溶剂2中与有机酸成盐纯化后的固体,加入二氯甲烷-20%液碱体系下解离后得到化合物Ⅳ;
3)化合物Ⅳ在溶剂3中通过10%钯碳,Raney-Ni,5%铂碳催化剂室温下氢解脱去氨基保护基并成磷酸盐,并通过丙酮-庚烷重结晶得到化合物Ⅰ。
所述化合物Ⅲ、化合物Ⅳ中R1基团为H或CH3或CH2CH3。
所述步骤1)手性路易斯催化剂为(S)-(-)-BINOL-Ti或(S)-(-)-BINOL-Mg络合物。
所述(S)-(-)-BINOL-Ti为摩尔比为1:1.8的S-BINOL和Ti(i-OPr)4制备的络合物,所述(S)-(-)-BINOL-Mg为摩尔比为1:1.4的S-BINOL和MgCl2制备的络合物。
所述步骤1)溶剂1为甲苯或二甲苯或四氯化碳,所述步骤1)中化合物为Ⅱ和化合物Ⅲ的摩尔比1:1.1,所述溶剂1是化合物为Ⅱ重量的9倍,所述手性路易斯酸催化剂为化合物为Ⅱ重量的0.12倍。
所述步骤2)溶剂2为丙酮,有机酸为马来酸。
所述步骤2)物料1与有机酸体积比为1:1.1,所述成盐纯化后的固体加入二氯甲烷与20%氢氧化钠体积比为2:1的溶液中,于28℃解离后得到化合物Ⅳ。
所述步骤3)中氢解使用的溶剂为乙醇。
所使用的催化剂为10%钯碳,Raney-Ni,5%铂碳,用量分别为化合物Ⅳ重量的2.6%、3.4%、1.6%。
所述步骤3)重结晶所采用的体系为丙酮-庚烷,丙酮与庚烷比例为1:6。
实施例5
(3R,4R,5S)-4-苄胺基-5-乙酰胺基-3(1-丙氧乙酯)-1-环己烷-1羧酸乙酯马来酸盐的合成
在100ml干燥的二氯甲烷中依次加入2.86g的(S)-BINOL,2.84g的Ti(O-i-Pr)4和2g 4A分子筛,回流反应两个小时得到催化剂S-BINOL-Ti,降至室温备用。称取29.5g的化合物II,在-20℃下依次加入干燥的甲苯300ml,11.8g的苄胺,上述催化剂S-BINOL-Ti,保温搅拌反应24h。中控显示原料反应完成,滤去分子筛,滤液加水萃取,有机层浓缩后加入240ml乙醇,12.7g马来酸搅拌析晶,所得固体加入二氯甲烷100mL,20%氢氧化钠50mL,25-30℃搅拌2h后分层,有机层洗涤两次后减压蒸干得到化合物IV 21.3g,收率41.1%。
实施例6
(3R,4R,5S)-4-苄胺基-5-乙酰胺基-3(1-丙氧乙酯)-1-环己烷-1羧酸乙酯马来酸盐的合成
在100ml干燥的二氯甲烷中依次加入2.86g的(S)-BINOL,0.95g的无水MgCl2和2g 4A分子筛,回流反应两个小时得到催化剂S-BINOL-Mg,降至室温备用。称取29.5g的化合物II,在-20℃下依次加入干燥的甲苯300ml,11.8g的苄胺,上述催化剂S-BINOL-Mg,保温搅拌反应24h。中控显示原料反应完成,滤去分子筛,滤液加水萃取,有机层浓缩后加入240ml乙醇,12.7g马来酸搅拌析晶,过滤后所得固体加入二氯甲烷100mL,20%氢氧化钠50mL,25-30℃搅拌2h后分层,有机层洗涤两次后减压蒸干得到化合物IV 19.7g,收率38.0%。
实施例7
(3R,4R,5S)-4-胺基-5-乙酰胺基-3(1-丙氧乙酯)-1-环己烷-1羧酸乙酯磷酸盐的合成
在高压反应釜中依次加入中间体IV 50g,10%的Pd/C催化剂2g,乙醇350ml,1ml冰醋酸,在0.5MPa的H2气压下,室温反应24h左右,滤去钯碳催化剂,减压蒸去约一半的反应液,在残液中加入磷酸成盐,室温下搅拌2小时后转冰浴搅拌1小时,过滤后减压干燥得到粗品36.8g,收率72.4%。
实施例8
(3R,4R,5S)-4-胺基-5-乙酰胺基-3(1-丙氧乙酯)-1-环己烷-1羧酸乙酯磷酸盐的精制
取上步反应粗品10g,加入丙酮40ml,在60℃下加热溶解,缓慢滴加庚烷200ml,保温回流搅拌2小时,缓慢降温至25℃,保温一小时,再降温至0℃下,保温一小时,过滤,50℃下减压干燥得到目标化合物6.8g,白色晶体,收率68%。
1H-NMR(600MHz,CDCl3)δ(ppm):0.90(6H,t,J=7.4Hz),1.26(3H,t,J=7.1Hz),1.51(2H,m),1.54(2H,m),1.96(3H,s),2.27(1H,m),2.77(1H,m),2.90(1H,dd,J=8.6,6.1Hz),3.39(1H,m),3.79(1H,m),4.04(1H,m),4.17(2H,m),6.44(1H,d,J=7.7Hz),6.82(1H,m);13C-NMR(150MHz,CDCl3)δ(ppm):9.7,14.4,23.6,26.0,26.5,29.7,48.7,54.3,61.0,78.2,81.7,129.6,136.1,166.5,170.3;MS(ESI):312(M+H)+
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!