一种含晶体的聚单硫代碳酸酯及其制备方法与流程
本发明属于高分子材料合成领域,具体涉及一种含晶体的聚单硫代碳酸酯及其制备方法。
背景技术:
聚硫代碳酸酯是一种具有优异光学性能、化学稳定性和良好重金属离子吸附能力的含硫聚合物,在高性能光导纤维制造、粘合剂和含重金属离子废水处理方面有广阔的应用前景。通常聚硫代碳酸酯由二硫醇与光气或氯仿在有机溶剂中反应制得,由于所用的原料光气、氯仿和溶剂等的毒害性,使该合成路线难有生存的价值。另一种合成聚硫代碳酸酯的方法是通过五元或六元环状的单、二和三硫代碳酸亚丙酯的开环聚合来实现。但这些五元环或六元环化合物通常是由二硫化碳和环氧化物经选择性偶合反应而得,因而实际是一种间接的由二硫化碳制备聚硫代碳酸酯的方法。
申请人曾采用氧硫化碳与环氧化物共聚合成聚硫代碳酸酯,如采用3,5-二叔丁基水杨醛亚胺席夫碱铬络合物和锌-钴双金属氰化络合物直接催化氧硫化碳与环氧化物交替区域选择性共聚得到聚单硫代聚碳酸酯(CN103275313A和CN103275314A),但所得的聚单硫代聚碳酸酯均不结晶,都是无定形的非晶聚合物,这是因为三元环醚单体开环后导致单硫代碳酸酯之间的连续CH2结构仅有2个,加之取代基的存在,共聚物难以通过链折叠而结晶;更为严重的是,氧硫化碳与环氧化物共聚容易发生氧硫交换反应,也导致共聚物不结晶。
之前所报道的涉及氧杂环丁烷和二氧化碳/二硫化碳共聚形成的聚碳酸酯/聚硫代碳酸酯均呈现无定形态结构。
一般而言,结晶聚合物具有综合的优异力学和热学性能。然而迄今绝大部分报道的可生物降解聚合物是不结晶的,因此在性能上一直难以媲美烯烃聚合物。少数几种典型的可生物降解的结晶性聚合物因熔点太低或太高而又难以应用或者加工,难以与聚烯烃竞争。例如聚己内酯是一种不含手性碳原子的生物降解的结晶性聚合物,但其熔点仅有50-60℃,很多场合难以单独应用。又如聚乳酸是一种典型的具有手性中心的结晶聚合物,熔点达170℃,然而其本身在高温下的易降解和水解特性,熔体加工极易发生降解,这也限制了其大量的应用。因此,在生物降解聚合物的合成领域中,需要有类似聚乙烯的结晶聚合物,且熔融温度和加工温度之间有较宽的窗口,才可能与聚烯烃相竞争。至今未见相关的报道。从合成角度来看,不依赖手性碳原子引起的结晶避免了昂贵的手性催化剂或单体的使用,在材料制备方面,又可以采用现有的加工设备加工结晶性聚合物。迄今也未见满足上述条件的生物降解的结晶性聚合物的报道。
技术实现要素:
针对以上描述的聚硫代碳酸酯不结晶和结晶难等问题,本发明提出了一种以氧硫化碳和氧杂环丁烷为主要反应物通过催化开环加聚合成含晶体的聚单硫代碳酸酯的制备方法,制备得到含有不对称单硫代碳酸酯链节、含晶体的聚单硫代碳酸酯。
一种含晶体的聚单硫代碳酸酯,共聚物的结晶为球晶结构,结晶度为13-85%、结晶温度为60~110℃、熔融温度为100~135℃、熔融热焓值于4~90J/g范围内可调节。
一种含晶体的聚单硫代碳酸酯的制备方法,具体包括以下步骤:
步骤1,将氧硫化碳、氧杂环丁烷、第三单体、链转移剂、催化剂以及助催化剂加入至干燥的高压反应釜中,在0~150℃于自生压力下本体或溶液共聚反应0.5~24小时;
步骤2,经纯化干燥后得到含晶体的聚单硫代碳酸酯。
在步骤1中,催化剂与氧杂环丁烷的摩尔比为1:50~5000;优选为1:100-500,在优选的范围内,共聚反应可以快速平稳地进行,得到中高分子量的产物,这一区间的聚合物往往具有综合的力学和热学性能。
在步骤1中,氧硫化碳与氧杂环丁烷的摩尔比为0.5~10:1;优选为2~5:1,与氧杂环丁烷等量或过量的氧硫化碳有利于共聚反应的快速进行,有利于氧杂环丁烷的完全转化,也有利于达到100%全交替共聚。
在步骤1中,助催化剂与催化剂的摩尔比为1:1。
在步骤1中,所述的第三单体为共聚物链结构和结晶性能的调节剂,可以为3-取代氧杂环丁烷、环氧化合物、环酸酐或环内酯;当使用第三单体时,第三单体与氧杂环丁烷的摩尔比为1~100:100;优选为1~50:100,一般来说,较少量的第三单体可以保证共聚物具有较高的结晶性能。
其中,3-取代氧杂环丁烷为3-甲基-3-苄氧基甲基氧杂环丁烷、3-氯甲基-3-甲基氧杂环丁烷、3-甲基-3-氧杂环丁烷甲醇、3-氧杂环丁酮、3,3-二甲基氧杂环丁烷、3-(烯丙氧基)氧杂环丁烷或3-溴氧杂环丁烷中的至少一种;
环氧化合物为环氧乙烷、环氧丙烷、C5-C20的α-氧化烯、烯丙基缩水甘油醚、1,2-环氧丁烷、环氧氯丙烷、环氧异丁烷、环氧环己烷、4-乙烯基环氧环己烷、氧化柠檬烯、环氧环戊烷、氧化苯乙烯或烷基取代氧化苯乙烯中的一种或多种;
环酸酐为马来酸酐、戊二酸酐、丁二酸酐、二甘醇酐或邻苯二酸酐等;所述的环内酯为丙内酯、丁内酯、戊内酯、己内酯、庚内酯、乙交酯、丙交酯或二甲基三亚甲基酯的至少一种;
在步骤1中,溶剂为三氯苯、邻二氯苯、间二氯苯、对二氯苯、二甲基甲酰胺等中的任意一种。
在步骤1中,所述的链转移剂为水、醇、有机羧酸、遥爪聚合物,遥爪聚合物的羟基或羧基为分子量200-5000g/mol的端基,羟基或羧基与氧杂环丁烷的摩尔比为1:1~50,进一步优选为1:25~50,相对较少的链转移剂有助于共聚物分子量的提高和链转移剂的全部转化。
在步骤1中,所述的催化剂为有机金属铬络合物;有机金属铬络合物为3,5-二叔丁基水杨醛亚胺席夫碱铬络合物,缩写为(Salen)CrX,其通式为:
式中,X为负一价阴离子,负一价阴离子为卤素阴离子、NO3-、CH3COO-、CF3COO-、CCl3COO-、ClO4-、N3-、邻-硝基苯酚氧、对-硝基苯酚氧、间-硝基苯酚氧、2,4-二硝基苯酚氧、2,4,6-三硝基苯酚氧、对甲基苯磺酸根、OH-、SH-或对甲基苯甲酸根中的任意一种;
R1和R2为氢原子、Cn~Cnn的烷基或Cm~Cmm的芳基;R1和R2是邻二胺取代的烷基或芳基衍生物,优选为乙二胺、邻苯二胺或环己二胺;例如R1=R2=H、R1=R2=CH3、R1=R2=C6H5、R1+R2=-(C4H4)-、R1+R2=-(CH2)4-、R1=CH3,R2=H或R1=C2H5,R2=H中的任意一种;
R3为H、C(CH3)3、CH(CH3)2、CH3、CH2CH3、OCH3、OCH2CH3、Cl、Br或NO2基团中的任意一种;
R4为H、C(CH3)3、CH(CH3)2、CH3、CH2CH3、OCH3、OCH2CH3、Cl、Br或NO2基团中的任意一种;
R5为H、C(CH3)3、CH(CH3)2、CH3、CH2CH3、OCH3、OCH2CH3、Cl、Br或NO2基团中的任意一种;
R6为H、C(CH3)3、CH(CH3)2、CH3、CH2CH3、OCH3、OCH2CH3、Cl、Br或NO2基团中的任意一种。
在步骤1中,所述的助催化剂为有机碱、季铵盐或季膦盐;为N-甲基咪唑、4-二甲氨基吡啶、双(三苯基正膦基)氯化铵、三苯基膦、三苯胺、十六烷基三甲基卤化铵、十二烷基三甲基卤化铵、十烷基三甲基卤化铵、1,8-二氮杂双环[5.4.0]十一-7-碳烯、1,5,7-二氮杂双环[5.4.0]十一-7-碳烯、四烷基卤化铵盐或四烷基卤化膦中的任意一种。
在步骤2中,所制得的聚单硫代碳酸酯的数均分子量为1~300kg/mol,分子量分布为1.1~5.0,单硫代碳酸酯链节含量≥99%,主链为全交替结构。
在大量实验研究的基础上,本发明通过一种新的交替共聚方法,实现了在较高聚合温度下完全抑制氧-硫交换反应,得到了结构明确且结晶性能可调的聚单硫代聚碳酸酯,其具有降解的硫代碳酸酯结构,又能如同高密度聚乙烯般结晶,并有较宽加工窗口,也具有高的强度和韧性。
相对于现有技术,本发明具有显著的有益的技术效果:
(1)本发明得到了新结构的聚单硫代碳酸酯,是通过氧杂环丁烷与硫氧化碳为主要单体,并调节共聚反应得到的。
(2)本发明得到的结构可控、全交替和链节结构不对称的聚单硫代碳酸酯,是通过双组分催化体系在较高的反应温度下得到的,且不产生氧硫交换反应,能够得到链结构规整的共聚物。
(3)共聚物可以快速结晶,和高密度聚乙烯的结晶性能类似,且结晶度可调节,同时也具有优异的力学性能。这些显著不同于任何一种已知的可生物降解的共聚物。
附图说明
图1为COS/OX共聚聚合产物的1H NMR谱图。
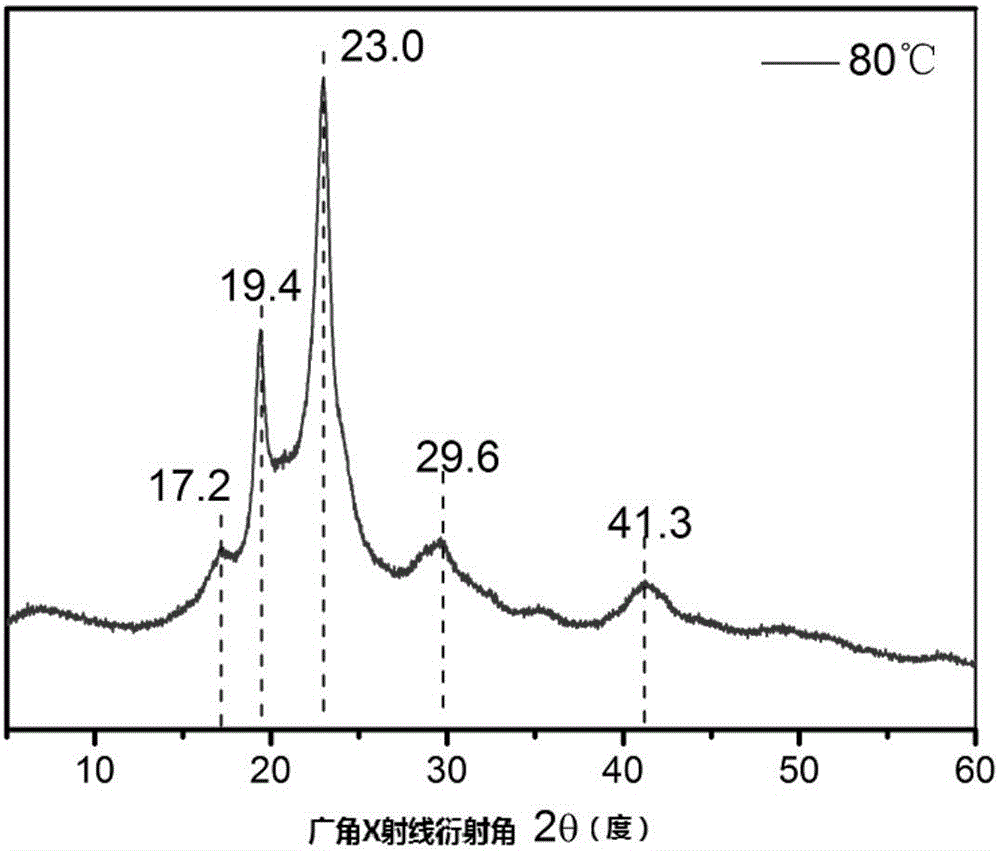
图2为COS/OX共聚得到的可结晶共聚物的X射线衍射图。
图3为COS/OX共聚得到的可结晶共聚物的偏光显微照片。
具体实施方式
为了更为具体地描述本发明,下面结合附图及具体实施方式对本发明的技术方案进行详细说明。
本发明中,所合成使用的催化COS/OX共聚合成聚单硫代碳酸酯所使用的催化剂具有以下结构,不同结构的催化剂序号如下式中所示。
实施例1(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温;依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数;催化剂与单体OX的摩尔比为1/250;再加入3.8g COS和2.0mL OX(COS与OX摩尔比2:1)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应4h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例2(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂1,5,7-二氮杂双环[5.4.0]十一-7-碳烯(TBD),二者等摩尔数。催化剂与单体OX的摩尔比为1/5000。再加入4.8g COS和1.0mL OX(COS与OX摩尔比5:1)。然后将高压反应釜封闭,置于150℃油浴中于自生压力下反应0.5h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例3(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1b和助催化剂十六烷基三甲基溴化铵(CTAB),二者等摩尔数。催化剂与单体OX的摩尔比为1/2500。再加入1.4g COS和3.0mL OX(COS与OX摩尔比0.5:1)。然后将高压反应釜封闭,置于0℃冰浴中于自生压力下反应24h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例4(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1c和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/500。再加入2.8g COS、0.3mL OX(COS与OX摩尔比10:1)和5mL三氯苯(TCB)。然后将高压反应釜封闭,置于80℃油浴中于自生压力下反应4h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例5(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1d和助催化剂4-二甲氨基吡啶(DMAP),二者等摩尔数。催化剂与单体OX的摩尔比为1/500。再加入1.4g COS和3.0mL OX(COS与OX摩尔比0.5:1)。然后将高压反应釜封闭,置于40℃油浴中于自生压力下反应24h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例6(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂2a(如图1)和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/50。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和5mL间二氯苯(MDCB)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例7(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂2b和助催化剂1,8-二氮杂双环[5.4.0]十一-7-碳烯(DBU),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和5mL二甲基甲酰胺(DMF)然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应10h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度。测试结果见表1。
实施例8(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂2c和助催化剂4-二甲氨基吡啶(DMAP),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS和2.0mL OX(COS与OX摩尔比2:1)。然后将高压反应釜封闭,置于100℃油浴中于自生压力下反应14h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度。测试结果见表1。
实施例9(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂2d和助催化剂1,5,7-二氮杂双环[5.4.0]十一-7-碳烯(TBD),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS和2.0mL OX(COS与OX摩尔比2:1)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应8h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度。测试结果见表1。
实施例10(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂3a和助催化剂1,5,7-二氮杂双环[5.4.0]十一-7-碳烯(TBD),二者等摩尔数。催化剂与单体OX的摩尔比为1/100。再加入3.8g COS和2.0mL OX(COS与OX摩尔比2:1)。然后将高压反应釜封闭,置于90℃油浴中于自生压力下反应15h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例11(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂3b(如图1)和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/2000。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和2.5mL邻二氯苯(ODCB)。然后将高压反应釜封闭,置于100℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例12(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂3c和助催化剂4-二甲氨基吡啶(DMAP),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS和2.0mL OX(COS与OX摩尔比2:1)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应10h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例13(Salen)CrX催化剂催化COS/OX一锅法共聚合成聚单硫代碳酸酯
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂3d和助催化剂十六烷基三甲基溴化铵(CTAB),二者等摩尔数。催化剂与单体OX的摩尔比为1/2500。再加入1.4g COS和3.0mL OX(COS与OX摩尔比0.5:1)。然后将高压反应釜封闭,置于70℃油浴中于自生压力下反应24h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,通过DSC测定聚合物的结晶温度,熔融温度以及熔融焓,通过X射线衍射测定聚合物的结晶度,测试结果见表1。
实施例14(Salen)CrX催化剂催化COS/OX/PO一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入7.6g COS、4.0mL OX(COS与OX摩尔比2:1)和2.25mL PO(PO与OX摩尔比1:2)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,测试结果见表2。
实施例15(Salen)CrX催化剂催化COS/OX/马来酸酐一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和0.06g马来酸酐(马来酸酐与OX摩尔比1:50)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,测试结果见表2。
实施例16(Salen)CrX催化剂催化COS/OX/己内酯一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a(如图1)和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和0.034mL己内酯(己内酯与OX摩尔比1:100)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
实施例17(Salen)CrX催化剂催化COS/OX/3,3-二甲基乙氧一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和3.34mL 3,3-二甲基乙氧(3,3-二甲基乙氧与OX摩尔比1:1)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布,测试结果见表2。
实施例18(Salen)CrX催化剂催化COS/OX/H2O一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和12μL去离子水(H2O与OX摩尔比1:50)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
实施例19(Salen)CrX催化剂催化COS/OX/甲醇一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和52μL甲醇(甲醇与OX摩尔比1:25)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
实施例20(Salen)CrX催化剂催化COS/OX/乙酸一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和1.85mL乙酸(乙酸与OX摩尔比1:1)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
实施例21(Salen)CrX催化剂催化COS/OX/甲氧基-PEG-丙酸一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和1.29g甲氧基-PEG-丙酸(Mn=2000g/mol)(甲氧基-PEG-丙酸与OX摩尔比1:50)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
实施例22(Salen)CrX催化剂催化COS/OX/聚己内酯一锅法共聚
聚合反应前先将10mL的高压反应釜于110℃下2小时左右除去水分并在干燥器中冷却至室温。依次向反应釜中加入若干质量的SalenCrX催化剂1a和助催化剂双(三苯基正膦基)氯化铵([PPN]Cl),二者等摩尔数。催化剂与单体OX的摩尔比为1/250。再加入3.8g COS、2.0mL OX(COS与OX摩尔比2:1)和3.22g聚己内酯(Mn=5000g/mol)(聚己内酯与OX摩尔比1:50)。然后将高压反应釜封闭,置于120℃油浴中于自生压力下反应12h。反应结束后冷却至室温,取出黄色产物。先用DMSO加热至130℃溶解粗产物,再在甲醇中沉淀出聚合物,重复洗三次得到白色产物,真空干燥至恒重。称重法计算转化率,通过核磁氢谱计算聚合物中各链节含量,通过凝胶色谱测定聚合物的分子量与分子量分布。测试结果见表2。
表1:实施例1-13的聚合产物的测试结果
1催化剂种类;2助催化剂种类。所加入助催化剂与催化剂等物质的量。[PPN]Cl=双(三苯基正膦基)氯化铵、DMAP=4-二甲氨基吡啶、CTAB=十六烷基三甲基溴化铵、DBU=1,8-二氮杂双环[5.4.0]十一-7-碳烯、TBD=1,5,7-二氮杂双环[5.4.0]十一-7-碳烯;3氧杂环丁烷与催化剂的摩尔比;4氧硫化碳与氧杂环丁烷的摩尔比;5ODCB:邻二氯苯,MDCB:间二氯苯,TCB:三氯苯,DMF:二甲基甲酰胺;6Mn:数均分子量,由凝胶渗透色谱法测定;7PDI:分子量分布,由凝胶渗透色谱法测定;8结晶温度,由差示扫描量热法测定;9结熔融温度,由差示扫描量热法测定;10熔融焓,由差示扫描量热法测定;11结晶度,由XRD估算。
表2:实施例14-21的聚合产物的测试结果1
1反应条件:催化剂为SalenCrX 1a,助催化剂为[PPN]Cl=双(三苯基正膦基)氯化铵,所加入助催化剂与催化剂等物质的量,催化剂与氧杂环丁烷的摩尔比为1:250,氧硫化碳与氧杂环丁烷的摩尔比为2:1,反应温度为120℃,反应时间为12h;2第三单体与氧杂环丁烷的摩尔比;3链转移剂与氧杂环丁烷的摩尔比;4Mn:数均分子量,由凝胶渗透色谱法测定;5PDI:分子量分布,由凝胶渗透色谱法测定;
由以上结果可知:
实施例1-13为(Salen)CrX催化剂搭配助催化剂催化COS/OX交替共聚的结果,可以看出(SalenCr)X催化剂具有优异的催化活性。在反应温度为90-120℃,COS/OX为2/1,反应时间4-12小时左右有较好的聚合结果,所得到的聚单硫代碳酸酯分子量达到19.1-300kg/mol;分子量分布为1.4-2.0;主链中不含有聚醚链节,单硫代碳酸酯链节比例≥99%。当在反应中添加溶剂时,如TCB或MDCB等,聚合物的分子量明显提高。当采用有机强碱作为助催化剂,如TBD或DBU,催化效率进一步提高,但所得聚合物的熔点和熔融焓均有所下降。
对采用本发明方法合成得到的产物进行结构分析,发现本发明的聚单硫代碳酸酯是一种结构新颖的含硫聚合物:在(Salen)CrX催化剂催化的氧杂环丁烷(OX)/COS共聚产物的1H NMR谱图(图1)上未见聚醚、聚硫醚以及其他各种硫代碳酸酯链节所对应的峰,说明所得产物为交替链节结构。单硫代碳酸酯链节上的各氢原子所对应的峰位置和积分面积如图1标注所示,从该图中可以看到副产物环状单硫代碳酸酯所对应的氢峰,粗产物中副产物和大分子的比例可以由4.47ppm和4.30ppm两处峰的面积比例所得。如图2所示,通过X射线衍射分析该聚单硫代碳酸酯材料在2θ=17.2°,19.4°,23.0°,29.6°,41.3°有强峰出现。图3为所得聚单硫代碳酸酯的偏光显微照片,从该图中可以看出所得共聚物的结晶含球晶结构。
将分子量为30kg/mol的聚单硫代碳酸酯热压成样条,其模量和断裂伸长率均优于同样分子量的高密度聚乙烯对比样品。
以上所述仅为本发明的若干个具体实施方式,应当指出,对于本领域的普通技术人员来说,还可以做出许多变型和改进,所有未超出权利要求所述的变型或改进均应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!