一种头孢替坦酸的新结晶及其制备方法与流程
本发明属于药物发明领域,具体涉及一种头孢替坦酸化合物的新结晶及其制备方法。
背景技术:
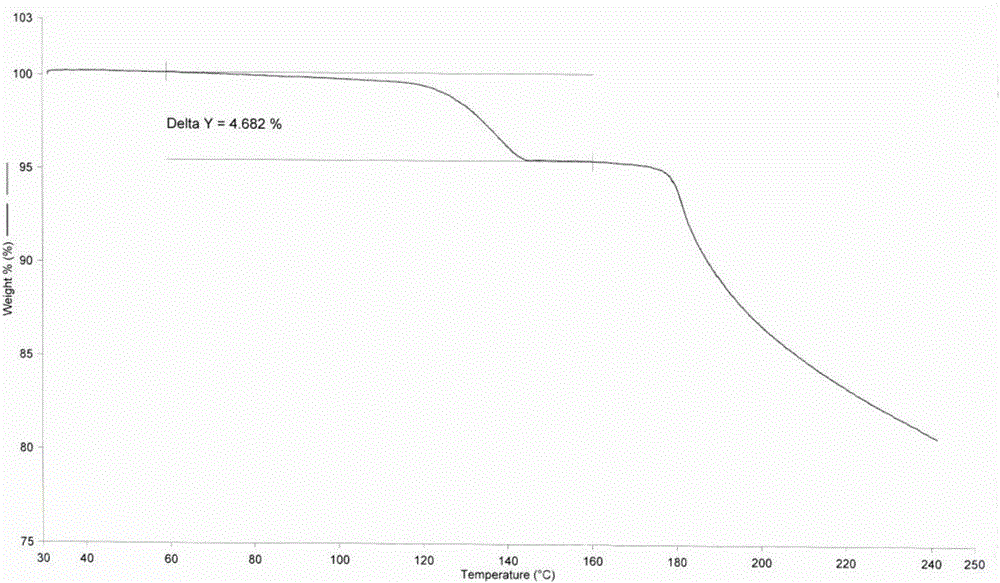
:头孢替坦酸(Cefotetan),化学名[6R-(6α,7α)]-7-[[[4-(2-氨基-1-羧基-2-氧代亚乙基)-1,3-二硫杂环丁烷-2-基]羰基]氨基]-7-甲氧基-3-[[(1-甲基-1H-四唑-5-基)硫代]甲基]-8-氧代-5-硫杂-1-氮杂双环[4,2,0]辛-2-烯-2-甲酸,是日本山之内制药开发的第二代头孢类抗生素,于1984年3月在日本上市,并载入日抗基。结构式如下:本品为头霉素类,作用与第三代头孢菌素近似。对各种细菌产生的内酰胺酶极为稳定,对革兰阴性菌和厌氧菌有良好的抗菌作用。特别是对大肠杆菌、沙雷菌属、变形杆菌属、摩根菌属、普罗威登斯菌属、假单细胞菌属及流感菌的抗菌作用比头孢美唑和头孢西丁强。但对革兰阳性菌作用不及一代与二代头孢菌素。临床主要用于呼吸道、肺部感染、腹部感染、尿路感染、妇科感染及中耳炎等。头孢替坦酸是一种常用的药物中间体,其极性较小,水溶性差,临床上为方便使用,以其钠盐制成制剂形式。但由于头孢替坦二钠很不稳定,在生产、储存和使用过程中遇水容易降解,有关物质会迅速增加,造成产品不合格,没办法使用,这给生产、储存和药品的安全使用都带来了极大的困难。现有技术中合成得到的头孢替坦酸如:专利申请CN103724359A中保护了一种无定形头孢替坦酸的制备方法。中国专利CN101050219A公开了一种头孢替坦二钠的制备方法,其中头孢替坦酸是在碳酸氢钠和盐酸中析出得到,CN101050219A报道了一种头孢替坦的制备方法,其中头孢替坦酸是在水相中析出得到。以上所述的现有技术合成得到了不同晶体结构的头孢替坦酸。本发明人在制备头孢替坦的过程中,通过溶媒析晶得到了一种不同于现有技术的新晶型的头孢替坦酸化合物,并通过验证,这种晶型的头孢替坦酸具有较好的稳定性,并且其制备工艺简单、制备成本更低、可工业化生产。技术实现要素:本发明的目的在于提供稳定性优良的一种头孢替坦酸的新型结晶,该晶型的X-射线粉末衍射图的反射角2θ在11.66±0.1°、17.53±0.1°、17.72±0.1°、18.91±0.1°、20.01±0.1°、21.02±0.1°、22.24±0.1°、23.33±0.1°处具有特征峰。本发明的另一目的在于提供一种所述的晶型头孢替坦酸的制备方法。为实现本发明的目的,本发明采用如下技术方案:本发明所述的晶型头孢替坦酸使用Cu-Kα射线测试得到X-射线粉末衍射图,如说明书附图图1所示。本发明所述的头孢替坦酸的晶型,其含有不少于1.5分子的结晶水,熔点为130±2℃,差示扫描量热吸收峰在120~165℃。如上所述晶型的头孢替坦酸具有更优的稳定性。本发明所述的一种头孢替坦酸新晶型的制备方法,该方法包括以下步骤:(1)将头孢替坦酸粗品溶解于2-丁酮和水的混合溶液中,得到头孢替坦酸的2-丁酮/水溶液;(2)向步骤(1)所得的头孢替坦酸的2-丁酮/水溶液中加入氯化钠,搅拌、静置、分层,取有机相;(3)将步骤(2)所得的有机相浓缩后,滴加无水乙醇,析晶、洗涤、过滤、真空干燥,得头孢替坦酸晶体。上述制备方法中,步骤(1)中所述2-丁酮和水的混合溶液中2-丁酮和水的质量分数之比为1:1,控制温度10~20℃。步骤(2)中所述搅拌和静置时间分别为15分钟。步骤(3)中所述搅拌析晶温度0~5℃,搅拌时间2小时,静置养晶温度-10℃,养晶时间10小时,真空干燥的温度为20~30℃。药物在结晶时,如果采用不同的溶剂和工艺,会形成不同的晶体结构,即药物的多晶现象。药物多晶型的变化会改变药物的性质、质量和药效。本发明对头孢替坦酸粗品进行重结晶,得到了一种新晶型的头孢替坦酸。通过验证表明本发明提供的头孢替坦酸具有较好的稳定性。由于本发明的头孢替坦酸新晶型可以直接将头孢替坦酸粗品反应获得,不需要从其他晶型转变而来,还具有操作简单、反应周期短、安全高效、制备成本较低的优点特别适合工业化生产。附图说明图1实施例2所得头孢替坦酸化合物的粉末X射线的衍射图;图2实施例2所得头孢替坦酸化合物的TGA分析图;图3实施例2所得头孢替坦酸化合物的DSC图;图4实施例2所得头孢替坦酸化合物的红外光谱图;图5实施例3所得头孢替坦酸样品放置0天的液相图;图6实施例3所得头孢替坦酸样品置于40℃放置5天的液相图;图7实施例3所得头孢替坦酸置于60℃放置5天的液相图;图8实施例3所得头孢替坦酸置于(4500±500)LX的照度下放置5天的液相图。具体实施方式除非另有定义,本发明使用的所有技术和科学术语的含义与本发明所属
技术领域:
普通技术人员通常理解的含义相同。通常,本发明使用的命名及实验操作方法都是本领域公知的或常用的,若未特别指明,本发明实施例中所用的试验试剂及材料均可市售获得。为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合具体实施例,对本发明作进一步的说明,而不是限制本发明。实施例1头孢替坦酸粗品制备(1)酰胺化反应将7-MAC用预冷的二氯甲烷溶解,加入反应釜中,搅拌冷却至-10℃以下,加入N,N-二甲基苯胺,搅拌10小时,滴加氯乙酰氯和二氯甲烷混合溶液,控温不高于-5℃。反应液用6%硫酸溶液洗涤两次,搅拌10分钟,分层,得有机层。再用冰水洗涤,静置分相,收集中间体I二氯甲烷溶液。(2)脱二苯酯反应将中间体I的二氯甲烷溶液加入反应釜中,冷却至5℃以下,加入正丁醇,搅拌15分钟后加入冷却过的三氟乙酸/苯甲醚溶液,0~5℃搅拌反应,直至反应结束,得反应液,待用。将反应液处理后,在30~35℃减压浓缩。向浓缩液中加入无水乙醇和异丙醇溶解,温度控制在0~10℃,滴加含异辛酸钠的异丙醇溶液,调节体系pH值至6.0±0.2,搅拌15分钟。滤饼用无水乙醇和异丙醇的混合溶液洗涤,继续离心至干,滤饼30~35℃真空干燥,得中间体II。(3)头孢替坦酸合成向反应釜中依次加入冷却过的纯化水,中间体II,开启搅拌。滴加5%碳酸氢钠溶液,调节pH值5.7~6.0,搅拌使固体完全溶解。然后按投料比依次加入EDTA二钠,碳酸氢钠。反应液通入CO2气体,并滴加三钠盐水溶液,控制反应体系pH值7.5~8.5,温度0~5℃。滴加完毕后,控制体系pH值7.9~8.1,反应8小时。然后通入CO2气体,调节反应液pH值7.5~7.8,HPLC监控异构化,直至异构体≤5.0%。滤液用9%盐酸水溶液调节体系pH至0.7~1.0,析出固体,离心过滤,得头孢替坦酸湿品。实施例2头孢替坦酸新晶型的制备将实施例1中得到的头孢替坦酸粗品加到2-丁酮和水的混合溶液中(W丁酮:W水=1:1),控制温度10~20℃,搅拌至固体完全溶解。向料液中加入氯化钠,搅拌15分钟,静置15分钟,分层,取有机相。有机相在20~30℃浓缩至约一半体积。浓缩完毕,浓缩液保持内温20~30℃,边搅拌边滴加无水乙醇。滴加完毕,降温至0~5℃,搅拌2小时使晶体析出,继续降温至-10℃,晶析熟化10小时,过滤。滤饼用乙醇洗涤,20~30℃真空干燥12小时,得头孢替坦酸,HPLC法含量测定:98.82%。所得头孢替坦酸结晶产品的X射线衍射、TGA、DSC和红外图谱如说明书附图1、图2、图3、图4所示。粉末X射线衍射测定条件:CuKα线,(单色管)、管电压40KV、管电流40mA。所得结晶的粉末X射线衍射测定结果示于图1中。就所得的结晶而言,X-射线粉末衍射图的反射角2θ在11.66±0.1°、17.53±0.1°、17.72±0.1°、18.91±0.1°、20.01±0.1°、21.02±0.1°、22.24±0.1°、23.33±0.1°处具有特征峰。此外,X-射线粉末衍射图的反射角2θ在9.89±0.1°、10.47±0.1°、11.42±0.1°、11.97±0.1°、13.31±0.1°、13.78±0.1°、14.25±0.1°、14.48±0.1°、16.36±0.1°、24.34±0.1°、25.88±0.1°、29.14±0.1°处有较低峰。表1是本发明头孢替坦酸新晶型X射线粉末衍射数据。表1:含水率:理论值(含1.5个水):4.48%热失重分析(TGA)测定值:4.682%差示扫描量热法(DSC)测得熔点:130.1℃将本发明的新结晶的红外光谱图如说明书附图4及下表所示。表2是本发明头孢替坦酸新晶型红外光谱数据。表2:实施例3头孢替坦酸新晶型的稳定性考察试验为了考察本品的热稳定性,将原料样品分置于40℃,60℃,(4500±500)LX的照度下影响30天,分别于第5、10、30天取样,考察外观性状、有关物质、含量变化,结果见表3。样品在0天,在40℃,60℃,(4500±500)LX的照度下分别放置5天的高效液相图谱如说明书附图5、图6、图7、图8所示。其中头孢替坦酸样品放置0天的液相检测图谱(即说明书附图5)中峰表如下:峰表检测器ACh1254nm峰#保留时间面积高度面积%分离度理论塔板#拖尾因子14.13439246670.0260.000101811.15625.11661728880.0415.598119721.02135.81735274120.0233.346100521.03547.7101188700.0086.21867080.78558.67524671190.0162.20148100.73169.37220551140.0141.49875511.090717.41382733230.05414.726111181.086820.4371508984326346399.3712.84531111.054924.940142353300.0943.51783051.1671027.4901197470.0083.030348770.6991144.218524684970.34612.98779211.435总计15185349266930100.000头孢替坦酸样品置于40℃放置5天的液相检测图谱(即说明书附图6)中峰表如下:峰表检测器ACh1254nm峰#保留时间面积高度面积%分离度理论塔板#拖尾因子16.4301118114880.0400.000154901.05927.14410891420.0043.420183150.95437.426915810890.0331.270163371.12249.33916771130.0066.593115240.885510.37449452740.0182.53978931.035611.28654962530.0201.86778341.118712.6542077730.0072.54279821.655820.08746162050.01713.026195550.926920.915155285820.0561.247123371.0341023.9972768456844273299.1892.65737511.0371129.138296036340.1063.75498920.9781231.762105502160.0382.578216191.1801337.254202722290.0733.97861680.7931450.7641102919610.3957.075111611.414总计27911051448990100.000头孢替坦酸样品置于60℃放置5天的液相检测图谱(即说明书附图7)中峰表如下:峰表检测器ACh1254nm峰#保留时间面积高度面积%分离度理论塔板#拖尾因子12.5892754929100.0830.00020981.40623.25757375080.0172.46316960.97733.58996000121890.2901.24544151.07143.91236454200.0111.42643661.08054.18636644170.0111.14147141.21165.66126821730.0084.74335901.14776.54412849652140.3881.73316620.93288.59831151290.0093.21129161.016910.225100613350.0302.31828441.0471012.8331600510.0053.93284101.5971114.1331421540.0042.24589340.0001216.3343256523257024198.2722.04517660.9231320.7047261111790.2192.68223660.9881423.083405466010.1221.33924881.1841526.462331064520.1001.87336311.0781638.33814223711510.4295.31732011.073总计33137703596022100.000头孢替坦酸样品置于(4500±500)LX的照度下放置5天的液相检测图谱(即说明书附图8)中峰表如下:峰表检测器ACh1254nm峰#保留时间面积高度面积%分离度理论塔板#拖尾因子16.3801423790.0180.00050520.70027.02615541500.0192.057108990.83737.65029562050.0371.82354451.18248.37030212050.0371.80575990.97659.7861083510.0133.17359260.704612.28117001040.0215.399139711.333714.823800720724173299.1384.00348191.001817.742168565330.2093.43670500.947919.39449922010.0622.218145031.0841022.5752844910.0354.122100781.1861125.3401618640.0203.555238701.9211230.459315564340.3912.73015511.279总计8076809243848100.000表3为本发明头孢替坦酸新晶体分别在高温、高湿、光照条件下的加速试验结果表3:测定结果发现,本发明的头孢替坦酸化合物在60℃放置30天后含量和有关物质的变化相对较明显,但在其它条件下头孢替坦酸化合物含量基本不变、有关物质无明显增加,且在限度要求范围内。实验结果说明本发明的头孢替坦酸化合物具有较好的储存稳定性。实施例4头孢替坦酸新晶型的溶解性考察试验根据头孢替坦酸合成工艺中用到的溶剂及质量检测可能用到的溶剂,我们选择了水、二氯甲烷、甲醇、乙醇、正丁醇、丁酮、乙酸乙酯作为溶剂,对头孢替坦酸的溶解度进行了研究。照《中国药典》2010年版二部凡例溶解度试验方法,称取研成细粉的供试品,置于25±2℃一定容量的溶剂中,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,如无目视可见的溶质颗粒,即视为完全溶解,结果见下表。表4为本发明头孢替坦酸新晶体分别在不同溶剂中的溶解情况表4:从表中可以看出,头孢替坦酸化合物在水、二氯甲烷、甲醇、正丁醇、乙酸乙酯中几乎不溶,在100mL乙醇中微溶,在100mL丁酮中溶解。结果表明本发明的头孢替坦酸的晶体稳定,不易发生变质。应当说明的是,虽然本发明以较佳实施例揭露如上,然其并非用以限定本发明,任何熟习此项技艺者,在不脱离本发明的精神和范围内,所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1