一种清洁抗污型两亲性共聚物网络及其制备方法与流程
本发明属于生物抗污材料领域,具体涉及一种新型的具有微相分离结构的两亲性共聚物网络的制备方法及其在生物医药领域的应用。
背景技术:
两亲性共聚物网络(Amphiphilic Conetworks,APCNs)自1988年被首次报道以来,目前已经成为一种具有广泛应用的生物高分子材料。它是由共价键互相连接两种具有连续形态的亲水链段和疏水链段(HI/HO),这两种链段分别聚集形成微相分离的相态结构,且各自保留原有的物理和化学性质,是一种具有介质(溶剂)响应性和形态异构化的“智能化聚合物网络”。广为人知的软性角膜接触镜就是一种由高度亲水相(通常为PDMAAm和PNVP)与高度疏水PDMS疏水相结合的APCN,且早在1971年,博士伦公司就已经将软性角膜接触镜(Soft Contact Lens)全面推向市场,视康公司也紧随其后推出了一系列以APCN型接触镜为主的软性染色及散光角膜接触镜。经过科学家们不断的发掘探索,这种具备良好生物相容性的两亲性共聚物网络的使用范围已经扩展到了药物缓释、医用分离、组织工程等领域。除此之外,APCN特殊的微相分离结构也有望应用在金属表面抗污粘合剂、智能响应新材料、高透氧膜制备等方面。
目前报道的APCN制备方法中主要有自由基聚合法、预聚物结合法以及活性可控聚合法。自由基聚合是一种由传统小分子单体与遥爪大分子单体进行自由基共聚制备APCNs的方法,也是目前合成APCN最广泛被使用的方法。根据自由基产生的方式可以分为热引发和光引发过程。热引发使用的是传统的偶氮与过氧化合物引发自由基生成。这种方法有个很大的优点就是简单,通过选用足够大相对分子质量的主链段(对于有效交联而言)和合成相对分子质量可精确控制的大分子交联剂,就可以利用共聚动力学将无规主链的相对分子质量精确控制并计算出来。然而热力学引发自由基聚合也有其缺点,例如会产生许多不可避免的副反应。光引发自由基聚合在合成APCNs方面尤其突出,如在角膜接触镜的应用领域上,被极其成功地用作长期佩戴型软性角膜接触镜的制备方法。例如,博士伦公司的Balafilcon A和视康公司的Lotrafilcon A两种角膜接触镜均是采用光交联/光引发共聚制备APCN的方法。德国学者Weber在1988年提出了化学偶联法制备APCNs,即分别合成功能化的亲水与亲油预聚物,再将其放入二者共同的良溶剂中。其简便高效,而且对目标产物的分子结构能起到很好的控制作用,通过控制预聚物的分子结构即可得到具有预期结构的APCNs体系,因此近年来发展很快。然而这种方法易产生悬垂链,降低网络机械性能。为了改善聚合可控性,提高网络力学性能,研究者提出了活性可控聚合法,通过精确控制嵌段长度从而连续制备两亲性嵌段共聚物,然后添加传统交联剂使其交联成具有统一筛孔尺寸的网络结构,现已经被系统地利用到了APCNs的合成中。其包括氮氧稳定自由基聚合(NMP)、原子转移自由基聚合(ATRP)、可逆加成-断裂链转移(RAFT)等。
构造APCN的难点在于控制亲疏水链段的分子量及其比例,以及提高交联固化的稳定性。因此必须要提高聚合过程的可控性以及交联反应的高效性,得到的交联网络不仅要有良好的机械性能,还要有足够的透明度和透氧性,而且无细胞毒性。
技术实现要素:
本发明的目的是提供一种具有清洁无污、良好抗蛋白吸附性的清洁抗污型两亲性共聚物网络及其制备方法。
为了达到上述目的,本发明提供了一种清洁抗污型两亲性共聚物网络,其特征在于,其制备方法包括:利用聚乙二醇制备亲水性大分子链转移剂;将所述的亲水性大分子链转移剂和疏水性单体进行RAFT聚合制备两亲性嵌段共聚物;通过胺解还原法除去所述的两亲性嵌段共聚物的末端三硫酯基;利用巯基-烯点击化学方法修饰和紫外光引发交联制备清洁抗污型两亲性共聚物网络。
优选地,所述的清洁抗污型两亲性共聚物网络的抗拉强度为1.5MPa~3.0MPa,断裂伸长率为50%~350%,在正己烷中溶胀度为20%~100%,在水中溶胀度为50%~300%,氧透过率为300~1000Barrers。
优选地,所述的清洁抗污型两亲性共聚物网络的溶胶含量不高于10%,抗拉强度为1.7MPa~3.0MPa,断裂伸长率为80%~350%。
本发明还提供了上述的清洁抗污型两亲性共聚物网络的制备方法,其特征在于,包括:
第一步:将RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、聚乙二醇、溶剂A、羧酸活化剂和脱水剂混合,在25℃~43℃下反应24~48小时,纯化,得到亲水性大分子链转移剂;
第二步:将所述的亲水性大分子链转移剂、疏水性单体、溶剂B和引发剂混合,在惰性气氛下,60℃~70℃下进行RAFT聚合反应8~24小时,纯化,得到两亲性嵌段共聚物;
第三步:取所述的两亲性嵌段共聚物、正胺、硫醇还原剂和溶剂C混合,在惰性气氛下,室温下反应10-60分钟后,注射添加丙烯酸酯,并反应10-24小时,纯化,得到通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物;
第四步:将第三步所得的两亲性嵌段共聚物同时进行巯基-烯点击化学方法修饰和紫外光引发交联,制备清洁抗污型两亲性共聚物网络;或将第三步所得的两亲性嵌段共聚物先进行巯基-烯点击化学方法修饰,得到氨基酸功能化的两亲性嵌段共聚物,再对氨基酸功能化的两亲性嵌段共聚物进行紫外光引发交联,制备清洁抗污型两亲性共聚物网络。
优选地,所述第一步中的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、聚乙二醇、溶剂A、羧酸活化剂和脱水剂的重量比为3.2∶5.8~16∶75~90∶0.3~0.5∶2.5~5.0。
优选地,所述第一步中的聚乙二醇的重均分子量为2k~10k,更优选为4k~6k,更优选为4k。
优选地,所述第一步中的羧酸活化剂为4-二甲氨基吡啶(DMAP),脱水剂为N,N’-二环己基碳二亚胺(DCC)或1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)。
优选地,所述第二步中的亲水性大分子链转移剂、疏水性单体、溶剂B和引发剂的重量比为4.5∶2.0~8.3∶85~95∶0.01~0.03。
优选地,所述第二步所得的两亲性嵌段共聚物中亲水链段与疏水链段的(重量比,除特别说明外,下文所述比例皆指重量比)比例为9/2~9/12,更优选为9/4~9/8。
优选地,所述第二步中的两亲性嵌段共聚物的重均分子量为8k~20k。
优选地,所述第二步中的疏水性单体为甲基丙烯酸烯丙酯(AMA)、丙烯酸丙炔酯或甲基丙烯酸丙炔酯,引发剂为偶氮二异丁腈(AIBN)、过氧化二苯甲酰(BPO)或偶氮(4-氰基戊酸)(ACVA)中的一种。
优选地,所述第三步中的两亲性嵌段共聚物、正胺、硫醇还原剂、溶剂C和丙烯酸酯的重量比为5.0∶0.1~0.7∶1.0~2.8∶92~95∶0.2~1.0。
优选地,所述第三步的催化剂为三乙胺TEA、正丁胺或偶氮二异丁腈AIBN中的一种。
优选地,所述第三步中的正胺为正丁胺,硫醇还原剂为(三(2-氯乙基)磷酸酯)(TCEP),丙烯酸酯为丙烯酸羟乙酯(HEA)或丙烯酸丁酯(t-BA)。
优选地,所述第四步中的对氨基酸功能化的两亲性嵌段共聚物进行紫外光引发交联的具体步骤包括:取氨基酸功能化的两亲性嵌段共聚物、交联剂、溶剂F和光引发剂混合,在紫外光照射下固化交联,得到清洁抗污型两亲性共聚物网络。
优选地,所述第四步中的利用巯基-烯点击化学方法修饰第三步所得的两亲性嵌段共聚物的具体步骤包括:将第三步所得的两亲性嵌段共聚物、氨基酸盐、溶剂D和催化剂混合,在惰性气氛下,在55℃~70℃下反应8~20小时,纯化,得到氨基酸功能化的两亲性嵌段共聚物。
优选地,所述第四步中的两亲性嵌段共聚物、氨基酸盐、溶剂D和催化剂的重量比为6.5∶0.8~3.6∶90~95∶0.01~0.05。
优选地,所述第四步的L-半胱氨酸盐酸盐和第二步的疏水性单体的比例为1/3~5/6,更优选为1/2。
优选地,所述第四步中紫外光的波长为365nm、功率4W,光强为0.5-1.5W/cm2,固化交联时间为20s~180s,更优选地,所述的光强为1.0W/cm2,固化交联时间为60s。
更优选地,所述第四步的光引发剂为安息香二甲醚(DMPA)光引发剂,所述的交联剂为(巯丙基)聚甲基硅氧烷(以下简称巯基硅油,生产厂家为美国GELEST公司,型号为SMS-992)。
优选地,所述的第四步中的将第三步所得的两亲性嵌段共聚物同时进行巯基-烯点击化学方法修饰和紫外光引发交联的具体步骤包括:将第三步所得的两亲性嵌段共聚物、氨基酸盐、溶剂D和催化剂混合,在惰性气氛下,在55℃~70℃下反应8~20小时,纯化,得到氨基酸功能化的两亲性嵌段共聚物。
更优选地,所述第四步的氨基酸功能化的两亲性嵌段共聚物、交联剂、溶剂F和光引发剂的重量比为10.0∶1.0~3.0∶85~93∶0.5。
优选地,所述第一步中的反应温度为36℃,第二步中的反应温度为63℃,第四步的反应温度为55℃。
优选地,所述的溶剂A、溶剂B、溶剂C、溶剂D和溶剂F独立地为二氯甲烷、四氢呋喃、1,4-二氧六环、正丁醇、甲苯、N,N-二甲基甲酰胺、二甲基亚砜的一种或两种以上的混合物。
优选地,所述的纯化包括用萃取剂萃取,萃取剂选择为正己烷、乙醚、石油醚,优选混合萃取剂正己烷/乙醚=1/1。
本发明还提供了上述的清洁抗污型两亲性共聚物网络在生物医药领域中的应用。
本发明还提供了上述的清洁抗污型两亲性共聚物网络在制备隐形眼镜、人工脏器和药物控制释放载体中的应用。
本发明采用可逆加成-断裂链转移聚合(Reversible Addition-Fragmentation Chain Transfer Polymerization,RAFT)对共聚物主链亲疏水链段的分子量进行可控聚合,得到含有亲疏水链段的两亲性共聚物,通过对末端进行胺解还原去色和利用巯基-烯点击化学(Thiol-ene Click Chemistry)对悬垂侧链进行半胱氨酸修饰,最后利用紫外光固化迈克尔加成交联制备出一种新型的两亲性抗污聚合物网络。
可逆加成-断裂链转移聚合是活性/可控自由基聚合(controlled radical polymerization,CRP)的一种,它是通过在聚合体系中加入链转移系数高的特种链转移剂,然后利用增长自由基与链转移剂的之间的可逆链转移反应(退化转移)来降低自由基的浓度,从而实现控制聚合体系中增长自由基浓度,达到活性可控的目的。RAFT法具有单体适用范围广、聚合条件温和(60~70℃下即可进行)、原料廉价易得、分子量可控且分子量分布较窄等优点,在实现分子设计的过程中,广泛用于合成一系列结构复杂、性能特殊的聚合物材料如嵌段、接枝、星状、梯状、超支化聚合物等,尤其是在制备网络尺寸可控的APCN方面,具有无法比拟的优势。
而通过RAFT活性可控聚合法合成的聚合物通常都带有活性官能团(C=S),这些活性官能团通常都具有一定的毒性,为达到更好的生物相容性,为后续构造具有良好生物相容性的生物材料,有必要对末端基三硫酯基进行还原去除。而利用简便的胺解还原反应,可以高效地去除三硫酯键,降低其生物毒性,即在RAFT聚合产物中添加少量的正胺,使其发生碳硫酯键的胺解还原反应,生成巯基(-HS)官能团,然后在TCEP试剂保护下,能继续与(甲基)丙烯酸(酰胺)发生迈克尔加成反应(Micheal addition reaction)。这种方法操作简便,且能有效去除二硫酯和三硫酯键。
点击化学是由诺贝尔化学奖得主Shapless在2001年提出的一个有机合成概念,它是指利用易得的化学原料,通过快速高效、高效的、具有选择性的模块化的化学反应来实现碳与杂原子之间的连接。典型的反应类型有铜催化的叠氮-炔基Husigen环加成反应和巯基-烯/炔点击反应。尤其是基于巯基无铜催化的烯/炔绿色点击反应,由于其具有反应条件简单、快速、强立体选择性、产物对水和氧不敏感、收缩应力低、产率高等优点,在基材表面修饰、纳米网络结构材料、聚合物功能化等方面被广泛研究。
本发明从绿色合成和原子经济角度出发,将以上技术高效结合制备出具有特定功能聚合物。具体地,采用RAFT技术制备亲水性大分子链转移剂,然后利用其可控聚合优势接入具有功能基团的疏水性丙烯酸酯,从而合成亲疏水链段及分子量可控的嵌段共聚物;接着通过添加少量正胺将末端三硫酯基胺解还原为巯基,而后在TCEP试剂保护下,继续与(甲基)丙烯酸(酰胺)发生迈克尔加成反应,形成稳定无毒的丙烯酸化末端;然后,结合巯基-烯点击化学在所合成聚合物悬垂侧链部分接入少量氨基酸类生理活性物质。最后,通过简单的迈克尔加成反应交联得到两亲性共聚物网络。
与现有技术相比,本发明的有益效果是:
本发明采用的聚合工艺条件温和、高效、可控性好,化学修饰方法清洁、简单,引发方式多样,交联所得的APCN具有性能优良、网络尺寸可控、力学强度、透明度高,韧性强,水油相中溶胀性能优良等优点,且热力学稳定性高于一般的线性高聚物,可广泛应用于药物缓释、隐形眼镜、生物导管等生物医用领域,同时在制备污涂层方面也具有很好的前景,可应用于海洋抗污涂层。
附图说明
图1为实施例2中原料PEG(a)、大分子链转移剂(b)和半胱氨酸修饰前后(c)&(d)的三嵌段两亲性共聚物的核磁共振谱图。从图中可以看出,(b)图在0.9ppm、1.8ppm、3.2ppm的化学位移出现了RAFT试剂末端峰,证明大分子链转移剂合成成功;(c)图在4.5-6.25ppm之间的化学位移处出现了代表碳碳双键氢的三个峰,证明了三嵌段制备成功;(d)图在4.2ppm和3.4ppm的化学位移处出现了半胱氨酸连接的α-碳氢和β-碳氢峰,证明半胱氨酸接入成功。
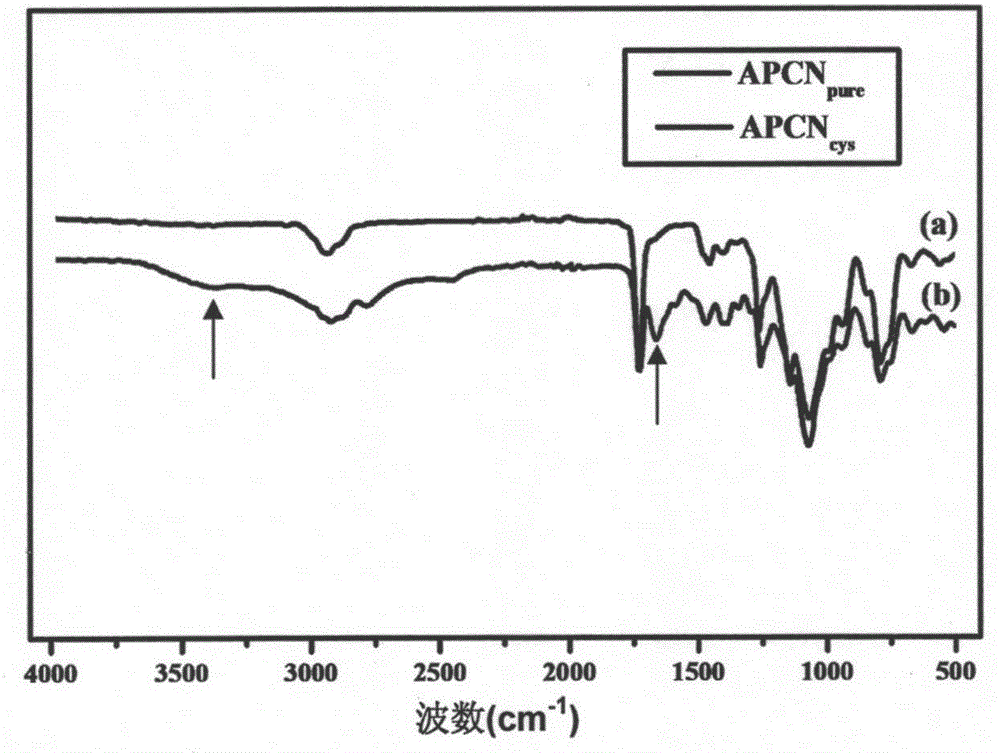
图2为实施例2中由两亲性嵌段共聚物在半胱氨酸修饰前后(a)&(b)制备得到的APCN的红外光谱。由图中可以看出,(a)中1648cm-1处不饱和碳碳双键吸收峰的消失说明了交联很充分,同时(b)图中在3380cm-1和1613cm-1处出现的吸收峰代表着半胱氨酸上氨基的伸缩振动和面内弯曲振动峰,而说明了对APCN半胱氨酸修饰成功。
图3为实施例2中所得两亲性共聚物网络(APCN)的原子力显微镜(AFM)相图。图中可看到明显的亲疏水相的连续形态,发亮区域为软段聚硅氧烷疏水链段,暗淡区域为硬段聚乙二醇亲水链段。相图结构无规地存在明显的界限,且为微纳米级别相态分离,这是APCN典型的微相分离结构。
图4为实施例2中两亲性嵌段共聚物在半胱氨酸修饰前(左)后(右)制备得到的APCN在荧光标记蛋白吸附测试中的激光共聚焦显微镜图。由图中可看到,纯APCN的蛋白吸附较为明显,且呈现团聚状态,而经半胱氨酸修饰后的APCN蛋白吸附量明显减少,且团聚不明显,说明制备得到的APCN具有比修饰前更好的抗蛋白吸附性。
图5分别为实施例2中所得两亲性共聚物网络的圆形干膜(左)和湿态膜(右)照片。得到的APCN均匀、无色透明,厚度为20-40μm,是一种柔软而富有弹性的纳米相膜材料。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
本文中涉及到多种物质的添加量、含量及浓度,其中所述的“份”,除特别说明外,皆指“重量份”;所述的百分含量,除特别说明外,皆指质量百分含量。
本发明的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸(TTC)]具体的合成方法如下:将250ml的三口烧瓶置于冰水浴(使瓶内温度小于10℃),通入氩气以排除瓶内空气后,依次加入聚四氟乙稀搅拌磁子、16.15g正十二烷基硫醇、48.5g丙酮、1.3g甲基三辛酰基氯化铵。混合溶液体系搅拌10min使其充分溶解后,通过恒压漏斗缓慢滴入6.67g质量分数为50%的氢氧化钠溶液,滴加完毕后持续搅拌15min。向体系中缓慢滴加含6.1g二硫化碳和6.8g丙酮的混合液,再次搅拌10min后,快速加入14.25g三氯甲烷。再次向反应瓶中缓慢滴加32g质量分数为50%的氢氧化钠溶液,20min内滴完,并室温下(25℃)搅拌过夜。向反应球瓶内加入120ml蒸馏水,然后滴加16-20ml浓盐酸使反应体系达到强酸性(PH~1),剧烈搅拌并通入氮气1h以除去残留的丙酮、氯仿、硫醇及二硫化碳等有机溶剂。过滤,取出上层固体并将其溶解于500ml异丙醇,再次过滤并取滤液,旋蒸浓缩至50ml左右。将得到的浓缩液趁热溶于200ml的正己烷中,于室温下(25℃)冷却,待粗产物结晶析出,过滤后取固体产物。重复重结晶操作(正己烷微热溶解-室温冷却结晶析出)以纯化产物。产物于50℃烘箱内真空干燥12h以上,最后得到淡黄色固体产物(产率为92.3%)(低温避光保存,熔点为60-63℃)。本发明中RAFT试剂也可采用市售产品,本发明其余原料为市售产品。
对下述各实施例得到的进行如下技术指标的测试评估。
一、测试方法及标准:
溶胶含量测试:将交联成膜后样品表面擦拭干净后称重,得初始质量m0,然后分别用DMF、甲苯、去离子水对膜样品洗涤,每种溶液均浸泡24小时。直至样品中未反应的溶胶全部被洗出。用试纸擦净并烘干后称重,得质量mt。按下式计算溶胶含量Sol%:
溶胀率(溶胀度)Sw测试:将干燥的膜样品称重,得初始质量m0,然后分别浸泡于去离子水、正己烷和乙醇中。在不同的时间点取样,用试纸擦净并烘干后称重,得质量mt,直到样品质量不再变化。下式为溶胀率(溶胀度)Sw%计算公式:
力学性能(抗拉强度、断裂伸长率)测试:将膜样品制成一定大小的条状,室温下用万能试验机(KEXIN,WDW3020,长春科新)测试。测试速率为10mm/min。每个样品至少测5次,以确保测量值的准确性。
氧透过率测试:使用OX2/23氧气透过率测试仪(济南兰光机电技术有限公司)对膜样品进行氧透过率测试。
表面粗糙度测试:利用原子力显微镜(Agilent 5500)观察膜表面形貌及粗糙度。轻敲模式,扫描范围:300nm×300nm。
蛋白吸附测试:制备异硫氰酸荧光素FITC标记的牛血清蛋白BSA溶液(FITC-BSA,0.01mol/L,PH=7.4)和PH7.4的磷酸盐缓冲液(PBS),每次以FITC-BSA∶PBS体积比为1∶10~1∶20稀释取用。测试时,将膜样品浸泡其中24小时后取出,用去离子水多次洗涤膜表面后用试纸擦干净表面残留的液体。采用激光共聚焦显微镜(*TCS SP5/TCS SP5 II)观察膜表面蛋白吸附情况。
二、实验材料:
1、RAFT试剂为自制,也可采用市售产品,制备过程见第8页。
2、巯基硅油是指(巯丙基)聚甲基硅氧烷,生产厂家为美国GELEST公司,型号为SMS-992。
其他试剂皆为分析纯,均购自中国医药(集团)上海化学试剂公司。
实施例1
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、0.3wt%的DMAP(4-二甲氨基吡啶)、2.6wt%的DCC(N,N’-二环己基碳二亚胺)、5.8wt%的PEG(聚乙二醇,Mw=2k)与88.1wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在室温下(25℃)磁力搅拌反应48小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃的冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步::将4.5wt%的PEG大分子链转移剂、8.3wt%的AMA(甲基丙烯酸烯丙酯)、0.01wt%的AIBN(偶氮二异丁腈)、87.19wt%的无水THF(四氢呋喃)混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于65℃油浴锅中,在氩气气氛下,进行RAFT聚合反应12小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为7.8kDa,其中,亲水链段与疏水链段的重复单元个数比为42∶40。
第三步:将5.0wt%的两亲性三嵌段共聚物、0.2wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%的两亲性嵌段共聚物、2.0wt%的L-半胱氨酸盐酸盐、0.01wt%的AIBN、91.49wt%的无水DMSO(二甲基亚砜)混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下反应18小时后,将混合体系转移至透析袋(MWCO=3.5-5kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.0wt%的巯基硅油、88.5wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.0W/cm2,固化交联时间为60s,得到清洁抗污型两亲性共聚物网络。
所得的清洁抗污型两亲性共聚物网络的抗拉强度为2.2MPa,断裂伸长率为103%,在正己烷中溶胀度为80%,在水中溶胀度为98%,氧透过率为200barrers,溶胶含量Sol=6.8%。核磁共振谱图、红外光谱图、AFM相图显示出实施例1得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例1制备的APCN的抗蛋白吸附情况(未图示)。
实施例2
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:利用聚乙二醇制备亲水性大分子链转移剂:将3.2wt%的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、0.4wt%的DMAP(4-二甲氨基吡啶)、2.6wt%的DCC(N,N’-二环己基碳二亚胺)、5.8wt%的PEG(聚乙二醇,Mw=4k)与88wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在室温下(25℃)磁力搅拌反应48小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将所述的亲水性大分子链转移剂和疏水性单体进行RAFT聚合制备两亲性嵌段共聚物:将4.5wt%的PEG大分子链转移剂、5.0wt%的AMA、0.01wt%的AIBN、90.49wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于65℃油浴锅中,在氩气气氛下,进行RAFT聚合反应12小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为10.0kDa,其中,亲水链段与疏水链段的重复单元个数比为90∶42。
第三步:通过胺解还原法除去所述的两亲性嵌段共聚物的末端三硫酯基:将5.0wt%的三嵌段共聚物、0.4wt%的正丁胺、痕量(0.001M%)的TCEP与94wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将所得的两亲性嵌段共聚物先进行巯基-烯点击化学方法修饰,得到氨基酸功能化的两亲性嵌段共聚物:将6.5wt%胺解还原后的三嵌段共聚物、1.4wt%的L-半胱氨酸盐酸盐、0.07wt%的AIBN、92.03wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下反应18小时后,将混合体系转移至透析袋(MWCO=3.5-5kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:对氨基酸功能化的两亲性嵌段共聚物进行紫外光引发交联,制备清洁抗污型两亲性共聚物网络:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.0W/cm2,固化交联时间为60s,得到清洁抗污型两亲性共聚物网络。
所得的两亲性共聚物网络的抗拉强度为2.5MPa,断裂伸长率为160%,在正己烷中溶胀度为52%,在水中溶胀度为180%,常温透光率为93%,溶胶含量Sol=4.1%。
图1为实施例2中原料PEG(a)、大分子链转移剂(b)和半胱氨酸修饰前后的三嵌段两亲性共聚物(c)&(d)的核磁共振谱图。从图中可以看出,(b)图在0.9ppm、1.8ppm、3.2ppm的化学位移出现了RAFT试剂末端峰,证明大分子链转移剂合成成功;(c)图在4.5-6.25ppm之间的化学位移处出现了代表碳碳双键氢的三个峰,证明了三嵌段制备成功;(d)图在4.2ppm和3.4ppm的化学位移处出现了半胱氨酸连接的α-碳氢和β-碳氢峰,证明半胱氨酸接入成功。
图2为实施例2中由两亲性嵌段共聚物在半胱氨酸修饰前后(a)&(b)制备得到的APCN的红外光谱。由图中可以看出,(a)中1648cm-1处不饱和碳碳双键吸收峰的消失说明了交联很充分,同时(b)图中在3380cm-1和1613cm-1处出现的吸收峰代表着半胱氨酸上氨基的伸缩振动和面内弯曲振动峰,而说明了对APCN半胱氨酸修饰成功。
图3为实施例2中所得两亲性共聚物网络(APCN)的原子力显微镜(AFM)相图。图中可看到明显的亲疏水相的连续形态,发亮区域为软段聚硅氧烷疏水链段,暗淡区域为硬段聚乙二醇亲水链段。相图结构无规地存在明显的界限,且为微纳米级别相态分离,这是APCN典型的微相分离结构。
图4为实施例2中两亲性嵌段共聚物在半胱氨酸修饰前(左)后(右)制备得到的APCN在荧光标记蛋白吸附测试中的激光共聚焦显微镜图。由图中可看到,纯APCN的蛋白吸附较为明显,且呈现团聚状态,而经半胱氨酸修饰后的APCN蛋白吸附量明显减少,且团聚不明显,说明制备得到的APCN具有比修饰前更好的抗蛋白吸附性。
图5分别为实施例2中所得两亲性共聚物网络的圆形干膜(左)和湿态膜(右)照片。得到的APCN均匀、无色透明,厚度为20-40μm,是一种柔软而富有弹性的纳米相膜材料。
实施例3
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、0.4wt%的DMAP(4-二甲氨基吡啶)、3.3wt%的DCC(N,N’-二环己基碳二亚胺)、11.2wt%的PEG(聚乙二醇,Mw=6k)与81.9wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在室温下(25℃)磁力搅拌反应48小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、3.4wt%的AMA、0.01wt%的AIBN、92.09wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于63℃油浴锅中,在氩气气氛下,进行RAFT聚合反应17小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为11.3kDa,其中,亲水链段与疏水链段的重复单元个数比为134∶36。
第三步:将5.0wt%的三嵌段共聚物、0.2wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原后的三嵌段共聚物、1.2wt%的L-半胱氨酸盐酸盐、0.06wt%的AIBN、92.24wt%的无水DMF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下反应18小时后,将混合体系转移至透析袋(MWCO=6-8kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.0W/cm2,固化交联时间为60s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为2.7MPa,断裂伸长率为113%,在正己烷中溶胀度为48%,在水中溶胀度为210%,氧气透过率为790barrers,溶胶含量Sol=8.4%。核磁共振谱图、红外光谱图、AFM相图显示出实施例3得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例3制备的APCN的抗蛋白吸附情况(未图示)。
实施例4
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、0.4wt%的DMAP(4-二甲氨基吡啶)、3.5wt%的DCC(N,N’-二环己基碳二亚胺)、16.0wt%的PEG(聚乙二醇,Mw=10k)与76.9wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在室温下(25℃)磁力搅拌反应48小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、2.1wt%的AMA、0.01wt%的AIBN、93.39wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于62℃油浴锅中,在氩气气氛下,进行RAFT聚合反应17小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为14.7kDa,其中,亲水链段与疏水链段的重复单元个数比为226∶37。
第三步:将5.0wt%的三嵌段共聚物、0.4wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.4wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、0.8wt%的L-半胱氨酸盐酸盐、0.05wt%的AIBN、92.65wt%的无水DMF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下反应18小时后,将混合体系转移至透析袋(MWCO=6-8kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.0wt%的巯基硅油、88.5wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.2W/cm2,固化交联时间为60s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为2.6MPa,断裂伸长率为65%,在正己烷中溶胀度为56%,在水中溶胀度为276%,氧气透过率为580barrers,溶胶含量Sol=9.1%。核磁共振谱图、红外光谱图、AFM相图显示出实施例4得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例4制备的APCN的抗蛋白吸附情况(未图示)。
实施例5
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂[2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸]、0.5wt%的DMAP(4-二甲氨基吡啶)、3.0wt%的DCC(N,N’-二环己基碳二亚胺)、8.6wt%的PEG(聚乙二醇,Mw=4k)与84.7wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在35℃磁力搅拌反应36小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、2.4wt%的AMA、0.01wt%的AIBN、93.04wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于70℃油浴锅中,在氩气气氛下,进行RAFT聚合反应8小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为7.2kDa,其中,亲水链段与疏水链段的重复单元个数比为90∶20。
第三步:将5.0wt%的三嵌段共聚物、0.2wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、0.9wt%的L-半胱氨酸盐酸盐、0.05wt%的AIBN、92.55wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于70℃油浴锅中,在氩气气氛下反应1小时后,将混合体系转移至透析袋(MWCO=3.5kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.5W/cm2,固化交联时间为60s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为2.1MPa,断裂伸长率为112%,在正己烷中溶胀度为60%,在水中溶胀度为143%,常温透光率为86%,溶胶含量Sol=8.9%。核磁共振谱图、红外光谱图、AFM相图显示出实施例5得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例5制备的APCN的抗蛋白吸附情况(未图示)。
实施例6
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂、0.4wt%的DMAP、2.5wt%的EDCI[1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐]、8.6wt%的PEG(Mw=4k)与85.3wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在35℃磁力搅拌反应36小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、3.6wt%的AMA、0.01wt%的AIBN、91.89wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于67℃油浴锅中,在氩气气氛下,进行RAFT聚合反应10小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为8.5,其中,亲水链段与疏水链段的重复单元个数比为90∶30。
第三步:将5.0wt%的三嵌段共聚物、0.4wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.4wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、1.1wt%的L-半胱氨酸盐酸盐、0.06wt%的AIBN、92.34wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于65℃油浴锅中,在氩气气氛下反应13小时后,将混合体系转移至透析袋(MWCO=3.5kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为0.8W/cm2,固化交联时间为180s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为1.8MPa,断裂伸长率为134%,在正己烷中溶胀度为87%,在水中溶胀度为101%,氧气透过率为565barrers,溶胶含量Sol=8.9%。核磁共振谱图、红外光谱图、AFM相图显示出实施例6得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例6制备的APCN的抗蛋白吸附情况(未图示)。
实施例7
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂、0.4wt%的DMAP、2.8wt%的EDCI、8.6wt%的PEG(Mw=4k)与85wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在43℃下磁力搅拌24小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、6.0wt%的AMA、0.02wt%的AIBN、89.48wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于63℃油浴锅中,在氩气气氛下,进行RAFT聚合反应17小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为11.0kDa,其中,亲水链段与疏水链段的重复单元个数比为90∶50。
第三步:将5.0wt%的三嵌段共聚物、0.6wt%的正丁胺、痕量(0.001wt%)的TCEP与94wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.4wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、1.8wt%的L-半胱氨酸盐酸盐、0.08wt%的AIBN、91.62wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下反应18小时后,将混合体系转移至透析袋(MWCO=3.5kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.0wt%的巯基硅油、88.5wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为0.6W/cm2,固化交联时间为180s,得到清洁抗污型两亲性共聚物网络。
所得的两亲性共聚物网络的抗拉强度为1.7MPa,断裂伸长率为163%,在正己烷中溶胀度为67%,在水中溶胀度为125%,氧透过率为367barrers,溶胶含量Sol=7.6%。核磁共振谱图、红外光谱图、AFM相图显示出实施例7得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例7制备的APCN的抗蛋白吸附情况(未图示)。
实施例8
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂、0.5wt%的DMAP、2.8wt%的EDCI、8.6wt%的PEG(Mw=4k)与84.85wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在43℃下磁力搅拌24小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、7.1wt%的AMA、0.03wt%的AIBN、88.37wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下,进行RAFT聚合反应24小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为12.2kDa,其中,亲水链段与疏水链段的重复单元个数比为90∶60。
第三步:将5.0wt%的三嵌段共聚物、0.6wt%的正丁胺、痕量(0.001wt%)的TCEP与94wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.4wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、2.3wt%的L-半胱氨酸盐酸盐、0.08wt%的AIBN、91.12wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于55℃油浴锅中,在氩气气氛下反应20小时后,将混合体系转移至透析袋(MWCO=6-8kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为0.5W/cm2,固化交联时间为115s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为1.9MPa,断裂伸长率为116%,在正己烷中溶胀度为78%,在水中溶胀度为123%,氧气透过率为327barrers,溶胶含量Sol=5.6%。核磁共振谱图、红外光谱图、AFM相图显示出实施例8得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例8制备的APCN的抗蛋白吸附情况(未图示)。
实施例9
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂、0.5wt%的DMAP、2.8wt%的EDCI、8.6wt%的PEG(Mw=4k)与84.9wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在室温下磁力搅拌48小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、5.0wt%的甲基丙烯酸丙炔酯、0.01wt%的AIBN、90.49wt%的无水DMF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于60℃油浴锅中,在氩气气氛下,进行RAFT聚合反应24小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为10.0kDa,其中,亲水链段与疏水链段的重复单元个数比为90∶42。
第三步:将5.0wt%的三嵌段共聚物、0.2wt%的正丁胺、痕量(0.001wt%g)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、2.3wt%的L-半胱氨酸盐酸盐、0.08wt%的AIBN、91.12wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于55℃油浴锅中,在氩气气氛下反应20小时后,将混合体系转移至透析袋(MWCO=6-8kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.2W/cm2,固化交联时间为95s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为2.2MPa,断裂伸长率为72%,在正己烷中溶胀度为87%,在水中溶胀度为110%,氧气透过率为423barrers,溶胶含量Sol=6.7%。核磁共振谱图、红外光谱图、AFM相图显示出实施例9得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例9制备的APCN的抗蛋白吸附情况(未图示)。
实施例10
一种清洁抗污型两亲性共聚物网络,其制备方法为:
第一步:将3.2wt%的RAFT试剂、0.5wt%的DMAP、4.0wt%的EDCI、16.0wt%的PEG(Mw=10k)与76.3wt%的无水二氯甲烷混合,并放入四氟搅拌磁子,在35℃下磁力搅拌36小时后,使混合物通过中性氧化铝层析柱(洗脱剂为二氯甲烷),将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后重新用二氯甲烷溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状PEG亲水性大分子链转移剂。
第二步:将4.5wt%的PEG大分子链转移剂、2.1wt%的AMA、0.01wt%的AIBN、93.39wt%的无水THF混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于65℃油浴锅中,在氩气气氛下,进行RAFT聚合反应12小时后取下冷却并通入空气,用10倍于产物量的0℃冰正己烷∶无水乙醚(1∶1)萃取洗涤纯化,并趁冷抽滤后重新用THF溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到淡黄色粉末状的两亲性三嵌段共聚物,重均分子量为14.7kDa,其中,亲水链段与疏水链段的重复单元个数比为226∶37。
第三步:将5.0wt%的三嵌段共聚物、0.2wt%的正丁胺、痕量(0.001wt%)的TCEP与94.2wt%无水THF混合,并放入四氟搅拌磁子。室温下(25℃)缓慢通入氩气,在氩气气氛下反应10分钟,溶液变成无色透明状,注射添加0.6wt%的t-BA(丙烯酸丁酯),继续反应10小时。将得到的滤液真空旋蒸浓缩,并用10倍于产物量的0℃冰正己烷萃取洗涤纯化,趁冷抽滤后用THF重新溶解。多次萃取溶解后,将产物于60℃真空干燥箱中烘干至恒重,得到白色粉末状通过胺解还原法除去末端三硫酯基的两亲性嵌段共聚物。
第四步:将6.5wt%胺解还原的三嵌段共聚物、0.8wt%的L-半胱氨酸盐酸盐、0.06wt%的AIBN、92.64wt%的无水DMSO混合,并放入四氟搅拌磁子。将反应体系置于低温恒温槽中冷却,维持内部温度5℃以下,向烧瓶中通氩气以排除空气,30分钟后密封体系并置于55℃油浴锅中,在氩气气氛下反应20小时后,将混合体系转移至透析袋(MWCO=6-8kD,美国光谱医学,再生纤维素)中纯化,外透析液为DMSO,共透析三次,每12小时换一次透析液。将透析液换成去离子水后实施同样的操作。冷冻干燥24小时后得到白色粉末状产物即半胱氨酸修饰的两亲性嵌段共聚物。
第五步:将10.0wt%经半胱氨酸修饰的两亲性嵌段共聚物、1.8wt%的巯基硅油、87.7wt%的DMF与0.5%的DMPA光引发剂混合均匀,并通过0.45μm的无纺布过滤,得到预交联溶液。将预交联溶液滴到载玻片上(四周由普通聚乙烯胶带围住,高度固定为1毫米),在紫外光照射下固化交联,所述的紫外光的波长为365nm、功率4W,光强为1.3W/cm2,固化交联时间为75s,得到清洁抗污型两亲性共聚物网络。
两亲性共聚物网络的抗拉强度为2.8MPa,断裂伸长率为62%,在正己烷中溶胀度为45%,在水中溶胀度为190%,氧气透过率为431barrers,溶胶含量Sol=9.7%。核磁共振谱图、红外光谱图、AFM相图显示出实施例10得到了两亲性共聚物网络;激光共聚焦扫描显微镜显示出实例10制备的APCN的抗蛋白吸附情况(未图示)。
- 还没有人留言评论。精彩留言会获得点赞!