一种合成聚醚酰亚胺的方法与流程
本发明涉及一种合成聚醚酰亚胺的方法,具体涉及半脂肪族、半脂环族或芳香族聚醚酰亚胺的合成方法,属于聚醚酰亚胺合成
技术领域:
。
背景技术:
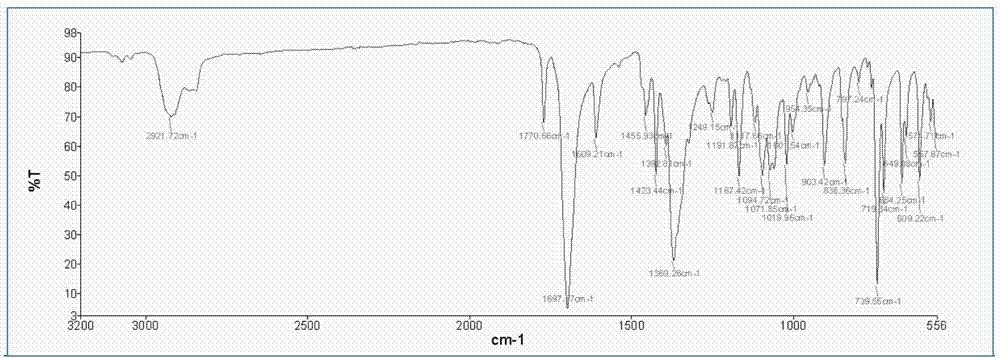
:聚醚酰亚胺是一种耐热且具有良好力学性能和加工性能的聚酰亚胺,该材料可以广泛地应用在轴承保持架、机械密封、烟草机械等领域。制备这一类材料,通常是先合成双醚四甲酸二酐,再由双醚四甲酸二酐与二胺在非质子极性溶剂中缩聚,化学亚胺化脱水得到聚醚酰亚胺。其中美国专利US5068353和中国公开专利CN104529966A中均介绍了双醚二酐的合成方法;在《聚酰亚胺—单体合成聚合方法及材料制备》中也详细介绍了聚醚酰亚胺树脂的制备方法以及材料性能。传统的聚醚酰亚胺合成方法是在非质子极性溶剂如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、N-甲基吡咯烷酮等中进行的。将二酐和二胺等摩尔进行缩聚形成聚醚酰胺酸,聚醚酰胺酸再经过去除溶剂、脱水亚胺化形成聚醚酰亚胺树脂或薄膜。美国专利文献US3179631和US3249588中均公开了由二酐和二胺在N,N-二甲基乙酰胺、N,N-二甲基甲酰胺或N-甲基吡咯烷酮中缩聚得到聚酰胺酸溶液,并将聚酰胺酸溶液加热或者加入叔胺催化剂加热亚胺化后得到聚酰亚胺的方法。反应过程如下:其中,R代表如等芳香族的基团。R'代表如等芳香族基团,R1代表如CH3-、C6H5-等烃基;X代表卤素。但传统的聚醚酰亚胺合成路线长、步骤多、成本偏高,并且目前极少见到脂环族或脂肪族的聚醚酰亚胺相关的学术报道。中国专利CN1560113中介绍了一种由双氯代双酞酰亚胺和二元酚在极性溶剂中缩合制备聚醚酰亚胺的方法,该方法明显具有路线短、成本低等特性。反应过程如下:其中,R代表如等芳香族的基团。R'代表如等芳香族基团。X代表卤素。此类反应的二胺原料都是常温下比较稳定、不易变质的二胺。但这一类二胺所制备的聚醚酰亚胺往往具有较强的刚性和较弱的溶解性,不适合制备一些性能特殊的聚醚酰亚胺。比如从理论上说,如果用甲基间苯二胺和双酚A二醚二酐合成的聚醚酰亚胺具有很好的溶解性,但是甲基间苯二胺在常温下不稳定,很容易被氧化,使得原料纯度降低,因此很难获得高分子量的“聚双酚A二醚四甲酰甲基间苯亚胺”这种材料。因此,需要寻找一种新的合成方法,从而制备出满足高分子量要求的“聚双酚A二醚四甲酰甲基间苯亚胺”。技术实现要素:本发明的目的是提供一种合成聚醚酰亚胺的方法,使用卤代苯酐、二异氰酸酯和二元酚作为原料,以六元杂环化合物作为催化剂制备得到满足高分子量、较好溶解性、并且常规方法不易合成的聚醚酰亚胺。为了实现以上发明目的,本发明采用的技术方案如下:一种合成聚醚酰亚胺的方法,包括以下步骤:a)卤代苯酐与二异氰酸酯在常温和惰性气体保护下,在非质子极性溶剂中反应生成双七元环物质;向其中加入六元杂环化合物作为催化剂,加热条件下双七元环物质进行脱二氧化碳的亚胺化反应,生成双卤代双酞酰亚胺溶液;b)在非质子极性溶剂中加入二元酚、无机碱性物质、液态烃和步骤a)得到的双卤代双酞酰亚胺溶液,在惰性气体保护下进行缩聚反应,当体系内无水分出时,继续反应若干小时后,蒸出液态烃,得到了聚醚酰亚胺溶液;c)步骤b)得到的聚醚酰亚胺溶液降温后,在搅拌下在醇类溶剂中析出,得到聚醚酰亚胺固体粉末。步骤a)中所述卤代苯酐结构式可表示为其中X代表卤素;所述卤代苯酐包括但不限于3-氯代苯酐、4-氯代苯酐、3-溴代苯酐和4-溴代苯酐等中的一种或多种。步骤a)中所述二异氰酸酯可表示为NCO-R″-NCO,其中,R”优选地代表脂环族或芳香族基团,如等;优选地所述二异氰酸酯是指碳原子数在8-15的脂环族和芳香族二异氰酸酯中的一种或多种,包括但不限于二环己基甲烷二异氰酸酯、异佛尔酮二异氰酸酯、二苯甲烷二异氰酸酯和甲苯二异氰酸酯中的一种或多种。步骤a)中所述卤代苯酐与二异氰酸酯的摩尔比为2:1。步骤a)中所述惰性气体包括但不限于氮气、氦气、氩气和氖气等中的一种或多种。步骤a)中所述非质子极性溶剂为沸点在150℃以上的非质子极性溶剂,包括但不限于N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲基亚砜、环丁砜和二苯砜中的一种或多种;步骤a)中所述卤代苯酐与二异氰酸酯的总量与步骤a)中非质子极性溶剂的质量比为15:100至25:100,优选18:100至20:100;举例说明,本发明通过研究发现甲基间苯二胺的光化产物甲苯二异氰酸酯的稳定性要比甲基间苯二胺好得多,因此如果能使用甲苯二异氰酸酯来合成双卤代双酞酰亚胺,之后再与双酚进行缩合反应,那么就可以以较短的路线来合成聚醚酰亚胺。本发明通过进一步研究发现:脂肪族酸酐(如乙酸酐)可以和异氰酸酯较快发生反应,生成如下基团:但由于苯酐、卤代苯酐等由于苯环的存在,在空间上使得酐基和异氰酸酯基反应速率明显偏慢。曾有文献报道,基团较复杂的酐基与异氰酸酯反应时间较长,通常需要20小时以上,且形成的七元环物质不易发生脱二氧化碳的反应,因此只能在反应体系中加入水、醇等物质作为脱二氧化碳的催化剂。但是作为本发明来将,如果在体系中加入水后,虽然反应可以顺利地进行,但是由于水的存在,会影响下一步的缩合反应的聚合度,那么如何快速地完成七元环脱二氧化碳成为本发明的一个重点。因此本发明中引入六元杂环化合物催化剂实现了加快反应时间的要求。本发明步骤a)反应过程如下所示:其中为步骤a)所述双七元环物质,为生成的双卤代双酞酰亚胺。Cat代表步骤a)所述催化剂六元杂环化合物,所述六元杂环化合物包括但不限于吡啶、甲基吡啶、喹啉、异喹啉、吖啶、哒嗪、嘧啶和吡嗪等中的一种或多种;催化剂的使用量为卤代苯酐质量的0.5-3%,优选1-1.5%。步骤a)中所述反应温度为60-150℃,优选70-120℃;反应时间为7-15小时,优选10-12小时。步骤b)中,所述非质子极性溶剂为沸点在150℃以上的非质子极性溶剂,包括但不限于N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲基亚砜、环丁砜、二苯砜中的一种或多种;优选地,步骤b)中,所述非质子极性溶剂与步骤a)中所述非质子极性溶剂的质量比为2.12:1至44.91:1,更优选2.46:1至32.61:1。步骤b)中,所述二元酚可表示为HO-R-OH,其中,R代表芳香族的基团,如等。优选地所述二元酚包括但不限于双酚A、双酚S、联苯二酚、萘二酚、对苯二酚和间苯二酚等中的一种或多种。优选地,步骤b)中所述二元酚的摩尔用量与步骤a)中所述二异氰酸酯相等。步骤b)中,所述无机碱性物质为能与二元酚反应生成二元酚盐的一元碱金属氢氧化物和/或碱金属碳酸盐,包括但不限于碳酸钾、碳酸钠、氢氧化钠和氢氧化钾等中的一种或多种,优选碳酸钾;所述无机碱性物质中碱金属离子与二元酚中酚羟基的摩尔比为1:1。步骤b)中,所述液态烃包括沸点110至180℃的脂肪烃和芳香烃中的一种或多种,包括但不限于正壬烷、正癸烷、十一烷、甲苯、二甲苯、乙苯和三甲苯等中的一种或多种;所述液态烃与步骤b)中所述非质子极性溶剂的质量比为1:3至1:5,优选1:3.5至1:4.5。步骤b)的反应过程如下所示:其中n代表聚合度。步骤b)中,其反应温度为160至220℃,优选180-200℃;反应时间为8-18小时,优选12-14小时。步骤b)中,二元酚和无机碱性物质的质量总和与液态烃和非质子极性溶剂的质量总和两者之间的比例为15:100至25:100,优选18:100至22:100。步骤b)中,所述惰性气体包括但不限于氮气、氦气、氩气和氖气等中的一种或多种。步骤c)中,所述聚酰亚胺溶液降温至80至155℃,优选90至150℃加入醇类溶剂。所述醇类为碳原子数1至5的水溶性低碳醇,包括甲醇、乙醇、异丙醇和异戊醇中的一种或多种,优选乙醇。本发明中,还包括对聚醚酰亚胺固体粉末进行后处理,包括过滤、洗涤、干燥,得到聚醚酰亚胺产品,所述聚醚酰亚胺为半脂环族或脂肪族聚醚酰亚胺。所述聚酰亚胺干燥方式为常压干燥,干燥时间为10至24小时,优选18-22小时。干燥温度为150至230℃,优选180-200℃。本发明的有益效果如下:1、采用二异氰酸酯作为制备聚醚酰亚胺的原料,解决了其对应二胺在常温条件下是极易变质,无法合成高分子量的聚醚酰亚胺的问题,从而合成出高分子量的聚醚酰亚胺。2、使用六元杂环化合物作为催化剂,加快了卤代苯酐与二异氰酸酯的反应速率,缩短了反应时间。3、本发明缩短了聚醚酰亚胺的合成步骤,使得合成过程简化。4、该合成方法无需进行化学亚胺化,解决了合成过程中带来的三废排放的问题。附图说明图1为实施例1合成的双氯代双酞酰亚胺的IR谱图图2为使用4,4’-二氨基二环己基甲烷与4-氯代苯酐为原料合成的双氯代双酞酰亚胺的IR谱图具体实施方式红外光谱仪:SHIMADAZUIRTracer-100,扫描范围400cm-1至4000cm-1。特性黏数测试标准:将0.5g树脂粉末加热溶于间甲酚中,定溶至100ml,之后在乌氏粘度计中测试特性粘数,单位dL/g:η=C-1ln(T/T’)C-1:溶液浓度;T:溶液流经圆球的时间;T'是甲酚流经圆球的时间。实施例1聚三苯二醚四甲酰二环己基甲烷亚胺的制备在带有搅拌的1000ml四口烧瓶中通入高纯氮气,加入250.8gN-甲基吡咯烷酮,开启搅拌,加入36.5g4-氯代苯酐粉末和26.2g经过纯化的二环己基甲烷二异氰酸酯。常温反应30min后,加入0.1825g吡啶,于120℃下反应15小时,当无二氧化碳气体溢出后,取样经液相色谱分析,当反应完全后,停止加热,得到了双氯代双酞酰亚胺溶液(进行红外分析,得到谱图详见附图1)。在1000ml三口烧瓶中加入101.8克二苯砜,加热至135℃融化后,通入氮气。在体系中同时加入11克对苯二酚、全部的双氯代双酞酰亚胺溶液、13.8克碳酸钾和78.31克正癸烷。待其全部均匀分布于体系中后,将体系升温至220℃。随着反应的进行,生成的水被带出反应体系。当反应18小时后,体系中无水分出,停止加热,降温至155℃后,将反应瓶内溶液倒入乙醇中,边搅拌边析出。将析出物经过过滤、乙醇洗涤和水洗后,将滤饼在230℃下进行干燥24小时,得到聚三苯二醚四甲酰二环己基亚胺粉末45.4克,收率93.8%。以间甲酚为溶剂测试其特性黏数为0.6822dL/g。从图1和图2可以看出,两个方法合成出来的双氯代双酞酰亚胺的IR谱图完全一致,证明了产物结构的一致。实施例2聚双酚S二醚四甲酰甲基间苯亚胺的制备在带有搅拌的1000ml四口烧瓶中通入高纯氩气,加入359.3gN-甲基吡咯烷酮,开启搅拌,加入36.5g4-氯代苯酐粉末和17.4g经过纯化的甲苯二异氰酸酯,反应30min后加入1.095g喹啉。120℃下反应15小时,当无二氧化碳气体溢出后,取样经液相色谱分析,当反应完全后,停止加热,得到了双氯代双酞酰亚胺溶液。在1000ml三口烧瓶中加入8克N-甲基吡咯烷酮,通入氩气。在体系中同时加入25.027克双酚S、全部的双氯代双酞酰亚胺溶液、13.8克碳酸钾和122.4克甲苯。待其全部均匀分布于体系中后,将体系升温至160℃。随着反应的进行,生成的水被带出反应体系。当反应8小时后,体系中无水分出,停止加热,降温至80℃后,将反应瓶内溶液倒入乙醇中,边搅拌边析出。将析出物经过过滤、乙醇洗涤和水洗后,将滤饼在150℃下进行干燥10小时,得到聚双酚S二醚四甲酰甲基间苯亚胺粉末57.8克,收率92%。以间甲酚为溶剂测试其特性黏数为0.5517dL/g。实施例3聚联苯二醚四甲酰二环己基甲烷亚胺的制备在带有搅拌的1000ml四口烧瓶中加入353g环丁砜,开启搅拌,在氩气保护下加入44.9g4-溴代苯酐粉末和26.2g(0.1mol)4,4’-二环己基二异氰酸酯(H12MDA),反应30min后加入0.449g哒嗪,保持反应体系70℃,反应10h后,当无二氧化碳气体溢出后,取样经液相色谱分析,当反应完全后,停止加热,得到了双溴代双酞酰亚胺溶液。在1000ml三口烧瓶中加入114.25克环丁砜,通入氮气。在体系中同时加入18.62克联苯二酚、全部的4,4’-双溴代双酞酰亚胺溶液、13.8克碳酸钾和116.8克邻二甲苯。待其全部均匀分布于体系中后,将体系升温至200℃。随着反应的进行,生成的水被带出反应体系。当反应14小时后,体系中无水分出,停止加热,降温至90℃后,将反应瓶内溶液倒入乙醇中,边搅拌边析出。将析出物经过过滤、乙醇洗涤和水洗后,将滤饼在200℃下进行干燥18小时,得到聚联苯二酚二醚四甲酰二环己基甲烷亚胺粉末60.0克,收率92%。以间甲酚为溶剂测试其特性黏数为0.5286dL/g。实施例4聚2,2’,3,3’-三苯二醚四甲酰异佛尔酮亚胺的制备在带有搅拌的1000ml四口烧瓶中加入263.5g二甲基亚砜,开启搅拌,在氮气保护下加入36.5g3-氯代苯酐粉末和22.2g异佛尔酮二异氰酸酯,反应30min后加入0.5475g甲基吡啶,保持反应体系60℃,反应12小时。当无二氧化碳气体溢出后,取样经液相色谱分析,当反应完全后,停止加热,得到了3,3’-双氯代双酞酰亚胺溶液。在1000ml三口烧瓶中加入138.6克环丁砜,通入氮气。在体系中同时加入11克对苯二酚、全部的3,3’-双氯代双酞酰亚胺溶液、8.0克氢氧化钠和80.42克乙苯。待其全部均匀分布于体系中后,将体系升温至190℃。随着反应的进行,生成的水被带出反应体系。当反应12小时后,体系中无水分出,停止加热,降温至120℃后,将反应瓶内溶液倒入异丙醇中,边搅拌边析出。将析出物经过过滤、异丙醇洗涤和水洗后,将滤饼在180℃下进行干燥22小时,得到聚3,3’,三苯二醚四甲酰异佛而酮亚胺粉末49.1克,收率91.6%。以间甲酚为溶剂测试其特性黏数为0.5419dL/g。实施例5聚3,3’,4,4’-三苯二醚四甲酰异佛尔酮亚胺的制备在带有搅拌的1000ml四口烧瓶中加入293.5gN,N-二甲基甲酰胺,开启搅拌,在氮气保护下加入36.5g4-氯代苯酐粉末和22.2g异佛尔酮二异氰酸酯,反应30min后加入0.438g异喹啉,保持反应体系90℃,反应11小时。当无二氧化碳气体溢出后,取样经液相色谱分析,当反应完全后,停止加热,得到了4,4’-双氯代双酞酰亚胺溶液。在1000ml三口烧瓶中加入9克环丁砜,通入氮气。在体系中同时加入11克对苯二酚、全部的4,4’-双氯代双酞酰亚胺溶液、8.0克氢氧化钠和86.43克邻二甲苯。待其全部均匀分布于体系中后,将体系升温至180℃。随着反应的进行,生成的水被带出反应体系。当反应13小时后,体系中无水分出,停止加热,降温至150℃后,将反应瓶内溶液倒入异丙醇中,边搅拌边析出。将析出物经过过滤、异丙醇洗涤和水洗后,将滤饼在190℃下进行干燥20小时,得到聚4,4’,三苯二醚四甲酰异佛而酮亚胺粉末49.1克,收率91.6%。以间甲酚为溶剂测试其特性黏数为0.5233dL/g。对比例1以非质子极性溶剂法为例,以3,3’,4,4’-三苯二醚四甲酸二酐和甲基间苯二胺为原料、N,N-二甲基乙酰胺为溶剂进行聚合,得到的聚三苯二醚四甲酰甲基间苯亚胺粉末进行测试其特性黏数为0.25dL/g。对比例2以非质子极性溶剂法为例,以3,3’-4,4’-双酚S二醚四甲酸二酐和甲基间苯二胺为原料、N,N-二甲基乙酰胺为溶剂进行聚合,得到的聚双酚S二醚四甲酰甲基间苯亚胺粉末进行测试其特性黏数为0.22dL/g。对比例3以非质子极性溶剂法为例,以3,3’-4,4’-联苯二酚二醚四甲酸二酐和4,4’-二氨基二环己基甲烷为原料、N,N-二甲基乙酰胺为溶剂进行聚合,得到的聚双酚S二醚四甲酰二环己基甲烷亚胺粉末进行测试其特性黏数为0.42dL/g。对比例4以非质子极性溶剂法为例,以2,2’,3,3’-三苯二醚四甲酸二酐和异佛尔酮二胺为原料、N,N-二甲基乙酰胺为溶剂进行聚合,得到的聚2,2’,3,3’-三苯二醚四甲酰异佛而酮亚胺粉末进行测试其特性黏数为0.24dL/g。对比例5以非质子极性溶剂法为例,以3,3’,4,4’-三苯二醚四甲酸二酐和异佛尔酮二胺为原料、N,N-二甲基乙酰胺为溶剂进行聚合,得到的聚3,3’,4,4’-三苯二醚四甲酰异佛而酮亚胺粉末进行测试其特性黏数为0.34dL/g。将本发明方法实施例1-5和非质子极性溶剂法的对比例1-5分别进行了特性黏数测试,详见表1。表1两种方法得到的聚醚酰亚胺的特性黏数比较序号特性黏数(dL/g)实施例对比例1聚三苯二醚四甲酰二环己烷甲烷亚胺0.68220.252聚双酚S二醚四甲酰甲基间苯亚胺0.55170.223聚联苯二酚二醚四甲酰二环己烷甲烷亚胺0.52860.424聚2,2’,3,3’-三苯二醚四甲酰异佛而酮亚胺0.54190.245聚3,3’,4,4’-三苯二醚四甲酰异佛而酮亚胺0.52330.34判断聚酰亚胺分子量是否足够大,是以0.5dL/g为标准,大于0.5dL/g,即可认为可以进行成型操作。从表中1可以看出,使用本发明方法得到的聚醚酰亚胺的分子量要大于传统的非质子极性溶剂法合成的聚醚酰亚胺的分子量。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1