一种2‑甲基‑3‑乙基马来酰胺制备方法与流程
本发明属于食品香料技术领域。更具体地,本发明涉及一种2-甲基-3-乙基马来酰胺制备方法。
背景技术:
马来酰胺类化合物广泛存在于葡萄、绿色植物等原料的天然降解产物中。研究表明,叶绿素A,在生物酶的作用下降解产生一系列马来酰亚胺类化合物。早在1948年叶绿素A的生物降解为马来酰亚胺类化合物过程已得到验证。Firmenish公司通过对烟叶提取及分析研究确定,烟草中含有大量的马来酰亚胺类化合物,并提出2-甲基-3-乙基马来酸酐对烟草的加香可能具有重要的积极意义。为进一步探讨酰亚胺类物质对食用及烟用加香的应用研究,本发明提出一种合成2-甲基-3-乙基马来酰亚胺的方法。
目前国内尚未有马来酰亚胺类化合物的合成方法,根据少量文献报道的半合成方法主要有通过叶绿素进行氧化后分离得到极低产率的目标产品。基于产品国内空白及产业化生产考虑,本发明涉及一种以工业原料马来酸酐为原料的合成工艺路线,通过加成及至酯胺解三步反应得到目标产品。
技术实现要素:
[要解决的技术问题]
本发明的目的是提供一种2-甲基-3-乙基马来酰胺制备方法。
[技术方案]
本发明是通过下述技术方案实现的。
本发明涉及一种2-甲基-3-乙基马来酰胺制备方法。
该制备方法的步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.25~0.5:2.5~4.5混合均匀,在温度100~120℃的条件下加热回流0.5h~4h,然后加入浓度为1~3mol/L硫酸水溶液,再加热回流0.5~4h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、干燥,得到所述的二甲基马来酸酐无色晶体;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:50~500:2.5~3.5,往步骤A得到的二甲基马来酸酐中添加四氯化碳与溴代试剂,混合均匀;所述的溴代试剂选自N-溴代丁二酰亚胺、N-溴乙酰胺、液溴、过溴化吡啶氢溴酸盐或二溴海茵;然后在温度80~100℃下加热回流2.5~5h,接着冷却至室温,经萃取、干燥、浓缩处理,得到一种溴代产物;按照溴代产物与四氢呋喃的重量比1:5~30,将溴代产物溶于四氢呋喃中;
按照溴代产物与催化剂I的摩尔比1:0.001~0.01,往溴代产物四氢呋喃溶液中添加亚铜化合物催化剂I,然后通N2气置换除去空气;
相继添加选自六甲基磷酰三胺、1,3-二甲基丙撑脲或1,3-二甲基-2-咪唑啉酮的催化剂II,溴代产物与催化剂II的摩尔比为1:10~20,同时将其温度降低至温度+5~-5℃;再缓慢滴加烷基格式试剂,溴代产物与烷基格式试剂的摩尔比为1:1.0~1.5,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再用乙酸乙酯萃取,有机相分离,干燥,浓缩,获得的粗产品经硅胶柱层析纯化,得到2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比为1:1.1~2.6,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度140~180℃,并在这个温度下反应1.5h~5h,然后冷却,升华,结晶,得到2-甲基-3-乙基马来酰胺白色针状晶体。
根据本发明的一种优选实施方式,在步骤A中,所述的干燥是在温度15℃~35℃下干燥30~60min。
根据本发明的另一种优选实施方式,在步骤B中,所述的萃取是溴化反应液与二氯甲烷萃取剂按照1:5~10重量比,在室温下混合萃取30~60min,按照同样方式萃取2~4次。
根据本发明的另一种优选实施方式,在步骤B中,萃取液在温度15℃~35℃的条件下进行干燥;在压力0.06~0.1KPa与温度20℃~25℃的条件下进行浓缩。
根据本发明的另一种优选实施方式,在步骤B中,亚铜化合物催化剂I是碘化亚铜、氯化亚酮或氧化亚铜。
根据本发明的另一种优选实施方式,在步骤B中,所述的烷基格式试剂选自甲基溴化镁、乙基溴化镁、正丙基溴化镁、异丙基溴化镁或正丁基溴化镁。
根据本发明的另一种优选实施方式,在步骤B中,乙酸乙酯萃取2-甲基-3-乙基马来酸酐粗品,反应液与乙酸乙酯萃取剂按照1:3~6重量比,在室温下混合萃取30~60min,按照同样方式萃取2~4次。
根据本发明的另一种优选实施方式,在步骤B中,有机相在温度15℃~35℃的条件下进行干燥;在压力0.06~0.1KPa与温度20℃~55℃的条件下进行浓缩。
根据本发明的另一种优选实施方式,在步骤B中,硅胶柱层析纯化采用填料为100~400目硅胶,溶剂选用石油醚、正己烷、乙酸乙酯。
根据本发明的另一种优选实施方式,在步骤C中,升华温度为140~160°,冷却介质选用空气、自来水、-10~10℃的冷却水。
下面将更详细地描述本发明。
本发明涉及一种2-甲基-3-乙基马来酰胺制备方法。2-甲基-3-乙基马来酰胺具有下述化学式:
该制备方法的步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.25~0.5:2.5~4.5混合均匀,在温度100~120℃的条件下加热回流0.5h~4h,然后加入浓度为1~3mol/L硫酸水溶液,再加热回流0.5~4h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、干燥,得到所述的二甲基马来酸酐无色晶体;二甲基马来酸酐具有下述化学式:
在本发明中,马来酸酐与冰醋酸的用量在所述范围内时,如果2-氨基吡啶的用量小于0.25,则马来酸酐原料残留;如果2-氨基吡啶的用量大于0.5,则收率不变,成本增加;因此,2-氨基吡啶的用量为0.25~0.50是合理的,优选地是0.30~0.45,更优选地是0.35~0.40。
同样地,马来酸酐与2-氨基吡啶的用量在所述范围内时,如果冰醋酸的用量小于2.5,则马来酸酐原料转化不完全;如果冰醋酸的用量大于4.5,则增加成本;因此,冰醋酸的用量为2.5~4.5是合理的,优选地是2.8~4.0,更优选地是3.2~0.36。
优选地,该反应在温度104~116℃的条件下加热回流1.2h~3.2h,更优选地,该反应在温度108~112℃的条件下加热回流1.8h~2.6h。
在这个步骤中添加硫酸水溶液的目的在于促进反应向产品推进。
在本发明中,如果硫酸水溶液的浓度小于1mol/L,则降低收率;如果硫酸水溶液的浓度大于3mol/L,则降低收率;因此,硫酸水溶液的浓度为1~3mol/L是可行的,优选地是1.4~2.6mol/L,更优选地是1.8~2.2mol/L。
在这个步骤中加热回流的主要作用是促进反应在0.5h~4h完成。加热回流的时间为0.5~4h,优选地是1.2~3.2h,更优选地是1.8~2.6h。
所述的干燥是在温度15℃~35℃下干燥30~60min,优选地是在温度18℃~32℃下干燥35~55min,更优选地是在温度23℃~28℃下干燥42~48min。
采用常规质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐。
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:50~500:2.5~3.5,往步骤A得到的二甲基马来酸酐中添加四氯化碳与溴代试剂,混合均匀;所述的溴代试剂选自N-溴代丁二酰亚胺、N-溴乙酰胺、液溴、过溴化吡啶氢溴酸盐或二溴海茵;然后在温度80~100℃下加热回流2.5~5h,接着冷却至室温,经萃取、干燥、浓缩处理,得到一种溴代产物;
在本发明中,二甲基马来酸酐与溴代试剂的用量在所述范围内时,如果四氯化碳的用量小于50,则容易形成二溴代副产物;如果四氯化碳的用量大于500,则二甲基马来酸酐反应不完;因此,四氯化碳的用量为50~500是合理的,优选地是120~380,更优选地是200~260。
同样地,二甲基马来酸酐与四氯化碳的用量在所述范围内时,如果溴代试剂的用量小于2.5,则二甲基马来酸酐反应不完;如果溴代试剂的用量大于3.5,则容易形成二溴代副产物;因此,溴代试剂的用量为2.5~3.5是恰当的,优选地是2.7~3.3,更优选地是2.9~3.0。
在本发明中,该溴代反应优选地在温度86~94℃下加热回流反应3.2~4.2h,更优选地在温度88~92℃下加热回流反应3.5~3.8h。
在萃取步骤中,溴化反应液与二氯甲烷萃取剂按照重量比1:5~10在室温下混合萃取30~60min,并且以同样方式萃取2~4次,得到的萃取液合并,接着在温度15℃~35℃的条件下进行干燥,以达到萃取液的水含量在以重量计5~8%以下,接着在压力0.06~0.1KPa与温度20℃~25℃的条件下浓缩直至合并萃取液体积的1/4~1/8,于是得到一种溴代产物。
按照溴代产物与四氢呋喃的重量比1:5~30,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.001~0.01,往溴代产物四氢呋喃溶液中添加亚铜化合物催化剂I,然后通N2气置换除去空气。在本发明中,添加亚铜化合物催化剂I的目的在于作为催化剂促进反应进行。所述的亚铜化合物催化剂I是碘化亚铜、氯化亚酮或氧化亚铜,
如果溴代产物与催化剂I的摩尔比大于1:0.001,则收率降低;如果溴代产物与催化剂I的摩尔比小于1:0.01,则产率不变,但增加成本;因此,溴代产物与催化剂I的摩尔比为1:0.001~0.01是恰当的,优选地是1:0.002~0.008,更优选地是1:0.004~0.006。
相继添加选自六甲基磷酰三胺、1,3-二甲基丙撑脲或1,3-二甲基-2-咪唑啉酮的催化剂II,溴代产物与催化剂II的摩尔比为1:10~20,同时将其温度降低至温度+5~-5℃;在本发明中,添加催化剂II的目的在于促进反应向目标产品进行。如果溴代产物与催化剂II的摩尔比大于1:10,则收率降低;如果溴代产物与催化剂II的摩尔比小于1:20,则产率不变,但增加成本;因此,溴代产物与催化剂I的摩尔比为1:10~20是恰当的,优选地是1:12~18更优选地是1:14~16。
本发明使用的催化剂II都是目前市场上销售的产品,例如由Sigma-Aldrich公司销售的六甲基磷酰三胺、由Sigma-Aldrich公司以商品1,3-二甲基-四氢-2-嘧啶酮销售的1,3-二甲基丙撑脲。
接着,缓慢滴加烷基格式试剂,溴代产物与烷基格式试剂的摩尔比为1:1.0~1.5,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;在本发明中,滴加烷基格式试剂的目的在于向溴代产物引入甲基。所述的烷基格式试剂选自甲基溴化镁、乙基溴化镁、正丙基溴化镁、异丙基溴化镁或正丁基溴化镁。本发明使用的烷基格式试剂是目前市场上销售的产品,例如由Sigma-Aldrich公司销售的甲基溴化镁。
在本发明中,如果溴代产物与烷基格式试剂的摩尔比大于1:1.0,则反应转化率降低;如果溴代产物与烷基格式试剂的摩尔比小于1:1.5,则反应完成但增加后处理的废水量;因此,溴代产物与烷基格式试剂的摩尔比为1:1.0~1.5是合理的,优选地是1:1.1~1.4,更优选地是1:1.2~1.3。
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:3~6,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取30~60min,按照同样方式萃取2~4次。反应液与乙酸乙酯萃取剂重量比为1:3~6是合理的,优选地是1:3.4~5.5,更优选地是1:4.0~4.8。
萃取完成后分离两相,合并的有机相在温度15℃~35℃的条件下进行干燥,在压力0.06~0.1KPa与温度20℃~55℃的条件下进行浓缩,获得2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚、正己烷或乙酸乙酯的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:1.1~2.6,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度140~180℃,并在这个温度下反应1.5h~5.0h,然后选用空气、自来水、-10~10℃冷却水的冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
在本发明中,如果2-甲基-3-乙基-马来酸酐与尿素的重量比大于1:1.1,则2-甲基-3-乙基-马来酸酐转化率降低;如果2-甲基-3-乙基-马来酸酐与尿素的重量比小于1:2.6,则尿素过量增加纯化难度;因此,2-甲基-3-乙基-马来酸酐与尿素的重量比为1:1.1~2.6是合理的,优选地是1:1.4~2.2,更优选地是1:1.6~2.0。
在这个步骤中冷却、升华与结晶使用的设备是化工技术领域里通常使用的设备。
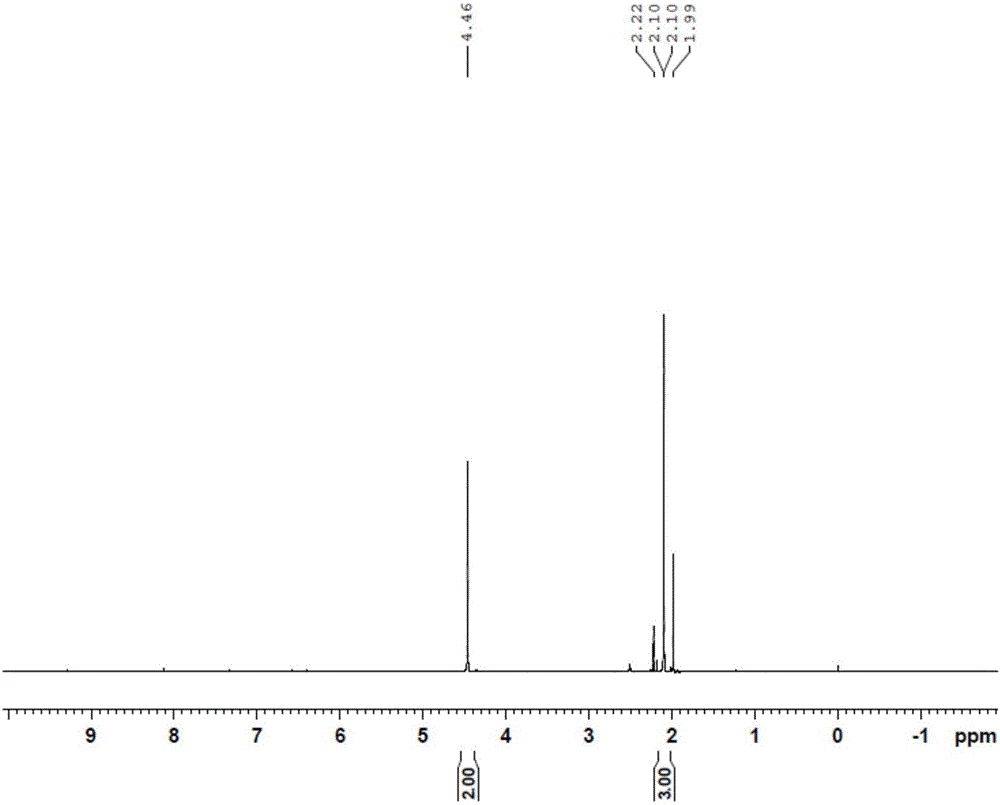
采用常规质子核磁共振分析方法,对本发明方法的不同步骤得到的产物无色晶体二甲基马来酸酐、2-甲基-3-乙基-马来酸酐、2-甲基-3-乙基马来酰胺进行了检测:根据分析检测结果确定得到的产物与设计产物结构一致。
其测定结果参见附图1-3。
根据质子核磁共振分析方法分析确定,本发明方法制备的2-甲基-3-乙基-马来酸酐纯度达到99%以上。
采用本发明方法制备2-甲基-3-乙基马来酰胺产物总收率通常达到8.6%。
[有益效果]
本发明的有益效果是:国内对马来酰亚胺类香料的研究基本空白。本发明的马来酰亚胺合成方法是以廉价易得的马来酸酐为原料,通过合成二甲基马来酸酐、2-甲基-3-乙基马来酸酐、再胺解后得到2-甲基-3-乙基马来酰胺。该工艺路线开发了一系列马来酰亚胺的合成方法,同时对这系列潜香物质进行加香应用实验,为开发新型香原料提供重要支持。
【附图说明】
图1是本发明实施例1制备的二甲基马来酸酐1H-NMR图。
图2是本发明实施例1制备的2-甲基-3-乙基马来酸酐1H-NMR图。
图3是本发明实施例1制备的2-甲基-3-乙基马来酰胺1H-NMR图。
【具体实施方式】
通过下述实施例将能够更好地理解本发明。
实施例1:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.25:2.8混合均匀,在温度104℃的条件下加热回流1.2h,然后加入浓度为1.8mol/L硫酸水溶液,再加热回流0.5h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度18℃下干燥42min,采用常规质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;该产物的质子核磁共振测定结果参见附图1。
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:120:2.5,往步骤A得到的二甲基马来酸酐中添加四氯化碳与N-溴代丁二酰亚胺溴代试剂,混合均匀;然后在温度88℃下加热回流3.5h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:8在室温下混合萃取54min,并且以同样方式萃取2次,得到的萃取液合并,接着在温度30℃的条件下进行干燥,以达到萃取液的水含量在以重量计5%以下,接着在压力0.06KPa与温度24℃的条件下浓缩直至合并萃取液体积的1/4,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:12,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.002,往溴代产物四氢呋喃溶液中添加碘化亚铜亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:10,相继添加六甲基磷酰三胺催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.4,缓慢滴加甲基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:4,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取30min,按照同样方式萃取4次,分离,合并的有机相在温度15℃的条件下进行干燥,在压力0.06KPa与温度50℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;该产物的核磁共振测定结果参见附图2。
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:1.8,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度140℃,并在这个温度下反应3.6h,然后选用空气冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。该产物的核磁共振测定结果参见附图3。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的总收率为6.5%。
实施例2:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.30:4.0混合均匀,在温度116℃的条件下加热回流3.2hh,然后加入浓度为2.2mol/L硫酸水溶液,再加热回流1.2h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度32℃下干燥48min,采用常规质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:380:2.7,往步骤A得到的二甲基马来酸酐中添加四氯化碳与N-溴乙酰胺溴代试剂,混合均匀;然后在温度92℃下加热回流3.8h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:5在室温下混合萃取46min,并且以同样方式萃取3次,得到的萃取液合并,接着在温度28℃的条件下进行干燥,以达到萃取液的水含量在以重量计6%以下,接着在压力0.08KPa与温度23℃的条件下浓缩直至合并萃取液体积的1/8,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:18,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.008,往溴代产物四氢呋喃溶液中添加氯化亚酮亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:20,相继添加1,3-二甲基丙撑脲催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.0,缓慢滴加乙基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:3,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取60min,按照同样方式萃取2次,分离,合并的有机相在温度18℃的条件下进行干燥,在压力0.08KPa与温度20℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂正己烷的常规硅胶柱层析纯化,得到的产物经常规核磁共振氢谱分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:1.4,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度160℃,并在这个温度下反应1.5h,然后选用自来水冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的总收率为6.8%。
实施例3:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.45:2.5混合均匀,在温度100℃的条件下加热回流0.5h,然后加入浓度为1.0mol/L硫酸水溶液,再加热回流3.2h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度15℃下干燥30min,采用核磁共振氢谱分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:50:3.3,往步骤A得到的二甲基马来酸酐中添加四氯化碳与液溴溴代试剂,混合均匀;然后在温度80℃下加热回流2.5h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:6在室温下混合萃取60min,并且以同样方式萃取4次,得到的萃取液合并,接着在温度15℃的条件下进行干燥,以达到萃取液的水含量在以重量计8%以下,接着在压力0.10KPa与温度20℃的条件下浓缩直至合并萃取液体积的1/6,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:5,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.001,往溴代产物四氢呋喃溶液中添加氧化亚铜亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:12,相继添加1,3-二甲基-2-咪唑啉酮催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.5,缓慢滴加正丙基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:5,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取40min,按照同样方式萃取3次,分离,合并的有机相在温度28℃的条件下进行干燥,在压力0.10KPa与温度55℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚、正己烷或乙酸乙酯的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:1.1,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度180℃,并在这个温度下反应4.2h,然后选用-10~10℃冷却水的冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的收率为6.52%
实施例4:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.5:2.5混合均匀,在温度120℃的条件下加热回流1.8h,然后加入浓度为1.4mol/L硫酸水溶液,再加热回流4.0h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度35℃下干燥60min,采用质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:200:3.5,往步骤A得到的二甲基马来酸酐中添加四氯化碳与过溴化吡啶氢溴酸盐溴代试剂,混合均匀;然后在温度100℃下加热回流5h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:8在室温下混合萃取30min,并且以同样方式萃取2次,得到的萃取液合并,接着在温度20℃的条件下进行干燥,以达到萃取液的水含量在以重量计5%以下,接着在压力0.06KPa与温度22℃的条件下浓缩直至合并萃取液体积的1/5,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:30,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.01,往溴代产物四氢呋喃溶液中添加碘化亚铜亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:18,相继添加1,3-二甲基丙撑脲催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.2,缓慢滴加异丙基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:6,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取50min,按照同样方式萃取2次,分离,合并的有机相在温度35℃的条件下进行干燥,在压力0.06KPa与温度40℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚、正己烷或乙酸乙酯的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:2.6,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度140℃,并在这个温度下反应5.0h,然后选用-10~10℃冷却水冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的总收率为6.55%
实施例5:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.40:3.5混合均匀,在温度108℃的条件下加热回流2.6h,然后加入浓度为2.6mol/L硫酸水溶液,再加热回流1.8h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度23℃下干燥35min,采用质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:260:2.9,往步骤A得到的二甲基马来酸酐中添加四氯化碳与二溴海茵溴代试剂,混合均匀;然后在温度86℃下加热回流3.2h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:10在室温下混合萃取38min,并且以同样方式萃取3次,得到的萃取液合并,接着在温度35℃的条件下进行干燥,以达到萃取液的水含量在以重量计6%以下,接着在压力0.08KPa与温度25℃的条件下浓缩直至合并萃取液体积的1/7,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:8,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.004,往溴代产物四氢呋喃溶液中添加氧化亚铜亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:16,相继添加1,3-二甲基-2-咪唑啉酮催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.3,缓慢滴加正丁基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:5,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取35min,按照同样方式萃取3次,分离,合并的有机相在温度22℃的条件下进行干燥,在压力0.08KPa与温度30℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚、正己烷或乙酸乙酯的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:2.2,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度180℃,并在这个温度下反应2.8h,然后选用空气冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的总收率为9.3%。
实施例6:2-甲基-3-乙基马来酰胺的制备
该实施例的实施步骤如下:
A、制备二甲基马来酸酐
马来酸酐、2-氨基吡啶与冰醋酸按照重量比1:0.40:4.5混合均匀,在温度112℃的条件下加热回流4h,然后加入浓度为3.0mol/L硫酸水溶液,再加热回流2.6h,冷却至室温,接着在搅拌下结晶,经过滤、水洗、在温度28℃下干燥55min,采用质子核磁共振分析方法检测确定,干燥得到的产物是无色晶体二甲基马来酸酐;
B、制备2-甲基-3-乙基马来酸酐
按照二甲基马来酸酐、四氯化碳与溴代试剂的重量比1:500:3.0,往步骤A得到的二甲基马来酸酐中添加四氯化碳与N-溴代丁二酰亚胺溴代试剂,混合均匀;然后在温度94℃下加热回流4.2h,接着冷却至室温,溴化反应液与二氯甲烷萃取剂按照重量比1:6在室温下混合萃取42min,并且以同样方式萃取4次,得到的萃取液合并,接着在温度24℃的条件下进行干燥,以达到萃取液的水含量在以重量计8%以下,接着在压力0.10KPa与温度23℃的条件下浓缩直至合并萃取液体积的1/5,于是得到一种溴代产物;
按照溴代产物与四氢呋喃的重量比1:25,将溴代产物溶于四氢呋喃中;按照溴代产物与催化剂I的摩尔比1:0.006,往溴代产物四氢呋喃溶液中添加碘化亚铜亚铜化合物催化剂I,然后通N2气置换除去空气。
按照溴代产物与催化剂II的摩尔比为1:15,相继添加六甲基磷酰三胺催化剂II,同时将其温度降低至温度+5~-5℃;接着,按照溴代产物与烷基格式试剂的摩尔比为1:1.2,缓慢滴加甲基溴化镁烷基格式试剂,在滴加过程保持其温度低于+5℃,并让该反应体系在该温度下保持过夜;
然后滴加饱和氯化铵水溶液使反应淬灭,再按照反应液与乙酸乙酯萃取剂重量比1:4,与烷基格式试剂反应所得到的反应液在室温下用乙酸乙酯混合萃取52min,按照同样方式萃取4次,分离,合并的有机相在温度24℃的条件下进行干燥,在压力0.10KPa与温度35℃的条件下进行浓缩,得到2-甲基-3-乙基马来酸酐粗产品,接着使用填料100~400目硅胶、溶剂石油醚、正己烷或乙酸乙酯的常规硅胶柱层析纯化,得到的产物经质子核磁共振分析检测确定是2-甲基-3-乙基-马来酸酐;
C、制备2-甲基-3-乙基马来酰胺
按照2-甲基-3-乙基-马来酸酐与尿素的重量比1:2.2,步骤B得到的2-甲基-3-乙基-马来酸酐与尿素搅拌混合均匀,再加热至温度180℃,并在这个温度下反应2.0h,然后选用自来水冷却介质进行冷却,再在升华温度140~160°下升华,结晶,得到白色针状晶体2-甲基-3-乙基马来酰胺。
由马来酸酐原料与白色针状晶体2-甲基-3-乙基马来酰胺的重量计算得到,本实施例的总收率为8.6%。
- 还没有人留言评论。精彩留言会获得点赞!
- 低介电常数树脂基材用处理铜箔及使用该处理铜箔的覆铜层压板以及印刷配线板的制造方法与工艺
- 含有色酮结构的吡唑类N‐对溴苯基马来酰亚胺衍生物及其制备方法与应用与制造工艺
- 一种双吲哚马来酰亚胺生物碱HD‑ZWM288在制备抗肿瘤微管抑制剂药物的应用的制造方法与工艺
- 马来酰亚胺膜的制造方法与工艺
- 多孔性酰亚胺系树脂膜制造系统、隔膜、及多孔性酰亚胺系树脂膜制造方法与制造工艺
- 聚苯并噁嗪前体及其制备方法与制造工艺
- 含酰亚胺结构的环三磷腈无卤阻燃剂、制备方法及用途与制造工艺
- 开关型硫醇荧光标记试剂及其合成方法和应用与制造工艺
- 一种耐高温表面自润滑的聚酰胺酰亚胺绝缘漆的制造方法与工艺
- 一种耐高温表面自润滑的聚酰胺酰亚胺绝缘漆的制造方法与工艺