N‑取代‑5‑亚甲基‑吡咯‑2‑酮类化合物的合成方法及应用与流程
本发明属于生物分子共轭标记领域,具体是N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法及其在可逆生物共轭标记中的应用。
背景技术:
带有标签和功能化基团的多肽和蛋白质被广泛用作药物分子或者生物医学研究的工具,而定点化学修饰是制备该类物质的重要手段[Chem.Rev.2015,115,2174-2195]。在20种天然的氨基酸中,半胱氨酸的天然丰度相对较低,且在生理条件下具有最高的亲核活性,因此是多肽和蛋白定点修饰最常用的氨基酸残基。传统的半胱氨酸特异性的生物共轭标记分子包括N-取代马来酰亚胺、2-碘代乙酰胺、卤代烷烃类和吡啶基二硫化物,如图1所示。
N-取代马来酰亚胺是目前应用最为广泛的半胱氨酸生物共轭标记分子。然而,它们也有其天然的缺陷[Bioconjugate Chem,2015,26,145-152]。首先,N-取代马来酰亚胺自身不稳定,易于水解开环而降解;其次,半胱氨酸-N-取代马来酰亚胺共轭产物也不稳定,一方面开环水解,另一方面能够和其他的巯基化合物发生交换反应。
最近,Kalia et al.报道了一种环外烯烃马来酰亚胺衍生物,能够高效、高选择性地对半胱氨酸进行共轭标记[Angew.Chem.Int.Ed.Engl.2016,55,1432-1435.],生成的共轭产物极其稳定。然而,在很多情况下需要可控地释放共轭功能基团。比如,通过氨基酸定点修饰制备的抗体偶联药物,在输送到靶细胞后,需要完整地将药物分子从修饰的氨基酸残基上释放出来,以发挥其药效[Angew.Chem.Int.Ed.2014,53,3796–3827]。再比如,组蛋白表观遗传修饰的添加及其移除与基因表达调控密切相关[Nat.Struct.Mol.Biol.2013,20,259-266],发展能够可控释放的蛋白标记新方法也能够为表观遗传研究提供新的工具。还有在蛋白质纯化及研究蛋白质相互作用方面,有时需要将靶蛋白固定在固相载体表面,最后施于温和的条件可以将蛋白完整地从载体表面洗脱下来。
目前文献报导的有一些半胱氨酸专一性、可控脱除的标记方法。比如吡啶基二硫化物和半胱氨酸共轭反应以后生成的二硫键可以在还原剂如巯基乙醇,谷胱甘肽存在的条件下断裂,释放出标记分子。还有Caddick等报道4-溴代哒嗪-3,5二酮与半胱氨酸的共轭产物也可以在过量的巯基乙醇存在的条件下释放出标记分子[Chem.Commun.,2011,47,8781–8783;J.Am.Chem.Soc.2012,134,1847-1852]。这些方法都利用高浓度的巯基小分子作为释放试剂,而巯基小分子本身可能和半胱氨酸反应,导致蛋白结构和功能的变化。因此发展新型的半胱氨酸高度专一性、更温和的条件下可控释放的蛋白标记试剂很有必要。
技术实现要素:
本发明的目的是针对上述存在问题,提供一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,制备的N-取代-5-亚甲基-吡咯-2-酮类化合物用于含巯基化合物、多肽和蛋白质的可逆标记以及在固相载体表面的固定。
本发明的技术方案:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为甘氨酰胺、荧光素、生物素、炔丙基、抗癌药物阿霉素或长链烷烃固相载体;
其合成方法,步骤如下:
1)将2,5-二甲氧基-2,5-二氢糠醇乙酸酯与0.1M的HCl水溶液按用量比1mmol:4mL混合,室温搅拌3h,用碳酸氢钠固体中和至中性,得到反应液;
2)在上述反应液中加入浓度为0.5M、pH为7.5的4-羟乙基哌嗪乙磺酸缓冲液至4-羟乙基哌嗪乙磺酸的终浓度50mM,得到混合液;
3)向上述混合液中加入含有伯氨基的化合物,混合液与含有伯氨基的化合物的用量比为5mL:0.5mmol,室温反应直至TLC板显示原料消失,旋干水溶液,粗产物用硅胶柱或反相HPLC分离纯化,制得N-取代-5-亚甲基吡咯-2-酮类化合物,所述含有伯氨基的化合物为甘氨酰胺、丙炔胺、生物素酰肼、阿霉素或氨基荧光素,含有伯氨基的化合物的化学结构式如式2所示,
4)向步骤2得到的混合液中加入长链烷烃氨基固相载体(LCCA-CPG),混合液与长链烷烃氨基固相载体的用量比为5mL:100mg,在室温下震摇4h,离心除去上清液,沉淀用去离子水洗涤两次、乙酸乙酯一次,最后用乙醚洗涤,自然晾干后,制得N-(LCCA-CPG)-5-亚甲基吡咯-2-酮。
一种所制备的N-取代-5-亚甲基吡咯-2-酮类化合物的应用,用于含巯基化合物、半光氨酸的可逆标记以及在固相载体表面的固定,方法如下:
1)将含巯基化合物与N-取代-5-亚甲基吡咯-2-酮类化合物加入pH 6.0的2-(N-吗啉代)-乙磺酸缓冲溶液中或pH 7.5的4-羟乙基哌嗪-乙磺酸缓冲液中,含巯基化合物与N-取代-5-亚甲基吡咯-2-酮类化合物用量的摩尔比为1:4-50,进行迈克尔加成反应生成5-取代-3-氢吡咯-2-酮,结构式如(3)所示:
2)将上述5-取代-3-氢吡咯-2-酮通过pH 9.5的硼酸钠缓冲溶液或者过量的巯基乙醇、谷胱甘肽处理,即可完整释放出被标记的半胱氨酸和巯基化合物。
本发明的优点是:
1)合成方法简单,原料易得,对大多数含伯氨基的化合物都能一步转化成标记分子;2)N-固相载体-5-亚甲基吡咯-2-酮标记在中性水溶液中很稳定;3)对巯基的选择性高,与巯基化合物的反应条件温和,反应速率快,产物结构单一,无手性中心生成;4)与巯基化合物的共轭交联产物在中性或弱酸性条件下稳定,在pH 9.5的缓冲溶液或过量巯基乙醇存在下,可以发生可逆反应消除标记分子,实现标记分子的可控释放。本发明所述的探针的优势,使其在蛋白质的可逆标记检测,抗体偶联药物的制备,蛋白质在固相载体表面的固定方面有着广泛的应用前景。
附图说明:
图1为传统的半胱氨酸生物共轭标记分子的结构。
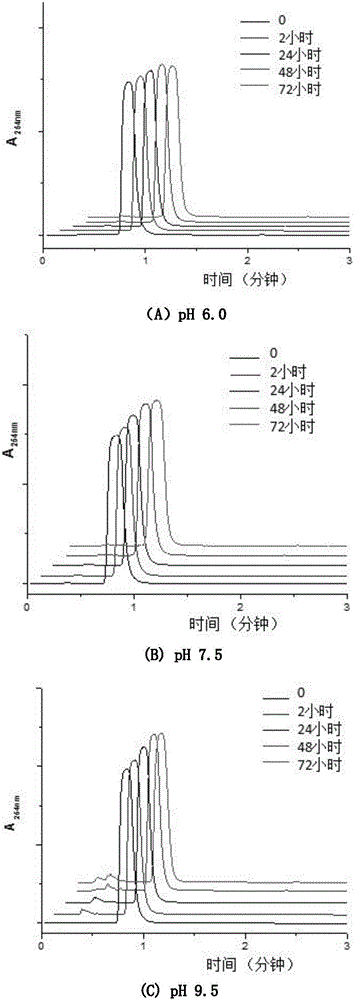
图2为UPLC分析化合物4分别在pH 6.0(A),7.5(B),9.5(C)的缓冲溶液中的稳定性。
图3(A)为5与谷胱甘肽GSH的巯基交换反应方程式;(B)为UPLC分析5与过量谷胱甘肽GSH的反应。
图4(A)标记分子4与多肽反应得到加成产物的结构;(B)为MS/MS分析确认分子4标记在半胱氨酸侧链上。
图5为MS分析标记分子4,7-10与蛋白质H4-R45C的反应。
图6为MS分析不同浓度的标记分子4与含有两个半胱氨酸的蛋白质ilvN的反应。
图7(A)为5-亚甲基-吡咯-2-酮修饰的固相载体从细胞裂解液中分离纯化ilvN-ilvB复合体示意图;(B)SDS-PAGE显示不同pH缓冲液从固相载体洗脱ilvN-ilvB复合体的效率;(C)SDS-PAGE显示SDS上样缓冲液(SDS LB)及700mM巯基乙醇从固相载体洗脱ilvN-ilvB复合体的效率;(D)比较pH 9.5缓冲液及700mM巯基乙醇从固相载体洗脱下来的ilvN-ilvB复合体的相对酶活性。
具体实施方式
以下对本发明的具体实施方式进行详细说明。此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
实施例1:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为甘氨酰胺;
合成方法如下:
称取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢糠醇乙酸酯于烧瓶中,加入4mL 0.1M的HCl水溶液,室温搅拌3h。反应体系用碳酸氢钠固体中和至中性,加入0.4mL、500mM的HEPES缓冲液调节pH至7.5。向得到的混合物中加入148mg(2mmol,2eq)的甘氨酰胺,室温反应1h。旋蒸除去溶剂,用甲醇/二氯甲烷(v/v,1/10)柱层析分离,得黄色固体136mg,产率90%。Rf=0.42(甲醇/二氯甲烷=1/10,v/v)。1H NMR(400MHz,DMSO)δ7.48(s,1H),7.31(d,J=5.6Hz 1H),7.15(s,1H),6.30(d,J=5.4Hz 1H),4.95(d,J=7.2Hz,2H),4.13(s,2H).13C NMR(101MHz,DMSO)δ170.1,169.4,145.8,138.8,124.8,97.5,41.5.HRMS(ESI):C7H8N2NaO2,[M+Na]+calc.175.0483,found 175.0491.
为了探究本发明设计合成的5-亚甲基吡咯酮类探针的稳定性,以标记分子(4)为研究对象,监测了(4)在不同pH缓冲溶液中的稳定性。探针分子(4)终浓度4mM,混合于100mM NaCl,20mM的MES,HEPES或硼酸-氢氧化钠缓冲液中,pH分别为6.0,7.5,9.5。37℃孵育。在不同的时间点取5μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图2表明,探针分子在不同pH水溶液中都很稳定,不会发生类似马来酰亚胺类探针的水解开环等副反应。
巯基乙醇与标记分子(4)的共轭加成反应:
称取152mg(1mmol,1eq)化合物(4)溶解于20mL HEPES(25mM,pH7.5)缓冲液中,均匀搅拌下加入280μL(4mmol,4eq)β-巯基乙醇。反应5min后,旋蒸除去溶剂,直接甲醇/二氯甲烷(v/v,6%)柱层析分离,得微黄色固体207mg(0.90mmol,产率90%)。Rf=0.29(甲醇/二氯甲烷:10%)。1H NMR(400MHz,DMSO)δ7.56(s,1H),7.18(s,1H),5.23(s,1H),4.86(t,3J=5.5Hz,1H),4.16(s,2H),3.56(dd,3J=6.4,12.8Hz 2H),3.42(s,2H),3.07(s,2H),2.58(t,3J=6.8Hz 2H).13C NMR(101MHz,DMSO)δ177.1,169.6,140.2,101.7,60.4,41.8,36.2,32.9,27.5;ESI-Q-TOF:C9H14N2NaO3S,[MNa]+cal.253.0623,found 253.0665。
共轭加成产物5的稳定性:
化合物5(终浓度4mM)溶于100mM NaCl,50mM的MES,HEPES,Tris或硼酸-氢氧化钠缓冲液中,pH分别为6.0,7.5,8.5,9.5,37℃孵育。在不同的时间点取样注射到UPLC-ESI-Q-TOF系统中检测反应情况。在中性pH7.5或者偏酸性pH6.0的条件下,标记产物相对稳定,其分解为初始探针的半衰期分别为21h和97h。在pH9.5下,共轭产物不稳定,逆释放出初始探针的半衰期为44min。
共轭加成产物5与巯基的交换反应:
化合物5(终浓度1mM)与20mM GSH混合于100mM NaCl,50mM的HEPES缓冲液中(pH7.5),37℃孵育。在不同的时间点取样注射到UPLC-ESI-Q-TOF系统中检测反应情况。图3显示5在22分钟内完全转变成新的共轭产物6,释放出巯基乙醇。
5-亚甲基吡咯-2-酮类标记分子4对多肽中半胱氨酸的特异性标记反应:
将多肽GCDWQSRMEYHTKN(终浓度0.4mM),与标记分子(4)(终浓度4mM)在37℃混合于100mM NaCl,20mM HEPES(pH7.5)缓冲液中,孵育1h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况,并用MS/MS确定探针标记位点。图4中表明,探针分子只特异性地标记多肽分子中的半胱氨酸。只得到单一的产物。
5-亚甲基吡咯-2-酮类分子4对蛋白质中单个半胱氨酸的特异性标记反应:
所采用的蛋白质为组蛋白H4的突变体H4-R45C,其45位含有一个半胱氨酸。其氨基酸序列如下:
SGRGKGGKGL GKGGAKRHRK VLRDNIQGIT KPAIRRLARR GGVKC45ISGLI
YEETRGVLKVFLENVIRDAVTYTEHAKRKT VTAMDVVYAL KRQGRTLYGF GG
将标记分子4(分子终浓度10mM)与组蛋白H4-R45C(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图5表明,标记分子5能高效地共轭标记到蛋白质上。
5-亚甲基吡咯-2-酮类分子4对含有多个半胱氨酸的蛋白质的标记反应:
采用的蛋白质为大肠杆菌AHAS I酶的小亚基ilvN,其含有两个半胱氨酸,氨基酸序列如下:
MGSSHHHHHHSSGLVPRGSHMQNTTHDNVILELTVRNHPGVMTHVCGLFARRAFNVEGILCLPIQDSDKS HIWLLVNDDQ RLEQMISQID KLEDVVKVQRNQSDPTMF NKIAVFFQ
将标记分子4(终浓度0至25mM)与ilvN(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH 7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图6表明,通过改变标记分子4的浓度,可以分别得到单、双共轭标记的产物。
实施例2:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为丙炔胺;
合成方法如下:
称取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢糠醇乙酸酯于烧瓶中,加入4mL 0.1M的HCl水溶液,室温搅拌3h。反应体系用碳酸氢钠固体中和至中性,加入0.4mL 500mM HEPES缓冲液(50mM,pH7.5)调节pH至弱碱性。向得到的混合物中加入28mg(0.5mmol,0.5eq)的炔丙基胺,室温反应1h。旋蒸除去溶剂,用石油醚/乙酸乙酯(v/v,4/1)柱层析分离,得无色油状液体54mg(产率80%)。Rf=0.49(石油醚/乙酸乙酯=4/1,v/v)。1H NMR(400MHz,CDCl3)δ7.01(d,J=5.2Hz,1H),6.19(d,J=5.6Hz,1H),5.12(s,1H),4.94(s,1H),4.40(s,2H),2.21(s,1H).13C NMR(101MHz,CDCl3)δ169.2,144.2,137.8,124.7,97.9,71.8,28.2;HRMS(ESI):C8H8NO,[M+H]+cal.134.0606,found 134.0636.
5-亚甲基吡咯-2-酮类分子7对蛋白质中单个半胱氨酸的特异性标记反应:
将标记分子7(分子终浓度10mM)与组蛋白H4-R45C(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图5表明,标记分子7能高效地共轭标记到蛋白质上。
实施例3:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为抗癌药物阿霉素;
合成方法如下:
称取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢乙酸糠醇酯于烧瓶中,加入4mL 0.1M的HCl水溶液,室温搅拌3h。反应体系用碳酸氢钠固体中和至中性,加入0.4mL 500mM HEPES缓冲液调节pH至7.5。向得到的混合物中加入290mg(0.5mmol,0.5eq)的阿霉素的DMSO(1mL)溶液,室温反应1h。用二氯甲烷5mL萃取三次,无水硫酸镁干燥,旋蒸除去溶剂,用甲醇/二氯甲烷(v/v,2%)柱层析分离,得红色固体125mg(0.20mmol,产率40.2%)。Rf=0.32(甲醇/二氯甲烷:2%)。1H NMR(400MHz,CDCl3)δ14.0(s,1H),13.3(s,1H),8.04(d,3J=7.6Hz 1H),7.81(t,3J=8.1Hz 1H),7.42(d,3J=8.4Hz 1H),6.98(d,3J=5.7Hz 1H),6.18(d,3J=5.7Hz 1H),5.87(s,1H),5.67(d,3J=3.2Hz 1H),5.41(br.,1H),5.36(t,3J=4.6Hz,1H),4.93(s,1H),4.90(m,1H),4.87(s,1H),4.80(s,2H),4.09(s,3H),3.98(s,1H),3.45(m,1H),3.31(d,3J=18.8Hz 1H),3.09(s,1H),2.83(m,1H),2.44(d,3J=14.7Hz,1H),2.02(m,1H),1.40(d,3J=6.4Hz 3H).13CNMR(101MHz,CDCl3)δ213.6,187.1,186.8,172.7,161.1,156.1,155.6,145.2,138.3,135.8,135.4,133.6,124.5,120.8,119.8,118.5,111.6,111.5,100.7,99.1,70.2,69.6,68.9,65.5,56.7,52.1,36.4,34.1,29.3,27.2,17.3.ESI-Q-TOF:C32H31NNaO12,[MNa]+cal.644.1744,found 644.1792。
5-亚甲基吡咯-2-酮类分子8对蛋白质中单个半胱氨酸的特异性标记反应:
将标记分子8(分子终浓度10mM)与组蛋白H4-R45C(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图5表明,标记分子8能高效地共轭标记到蛋白质上。
实施例4:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为荧光素;
合成方法如下:
称取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢乙酸糠醇酯于烧瓶中,加入4mL 0.1M的HCl水溶液,室温搅拌3h。反应体系用碳酸氢钠固体中和至中性,加入0.4mL 500mM HEPES缓冲液调节pH 7.5。向得到的混合物中加入174mg(0.5mmol,0.5eq)的5-氨基荧光素的甲醇(1mL)溶液,室温反应1h。旋蒸除去溶剂,直接用甲醇/二氯甲烷(v/v,2%)柱层析分离,得黄色固体88mg(0.21mmol,产率41.4%)。Rf=0.35(甲醇/二氯甲烷:2%)。1H NMR(400MHz,DMSO)δ10.46(br.,2H),7.88(s,1H),7.71(d,3J=7.9Hz,1H),7.60(d,3J=4.6Hz,1H),7.40(d,3J=8.0Hz,1H),6.78(s,2H),6.63(q,3J=8.6Hz,22.8Hz,4H),6.48(d,3J=4.6Hz,1H),5.21(s,1H),5.12(s,1H).13CNMR(101MHz,DMSO)δ207.9,169.5,168.4,160.3,152.3,151.2,145.6,139.9,136.1,135.0,132.1,130.2,130.1,129.6,127.9,125.5,124.3,123.6,113.3,109.6,102.8,100.1,93.8.ESI-Q-TOF:C25H16NO6,[MH]+cal.426.0978,found 426.1007。
5-亚甲基吡咯-2-酮类分子9对蛋白质中单个半胱氨酸的特异性标记反应:
将标记分子9(分子终浓度10mM)与组蛋白H4-R45C(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图5表明,标记分子9能高效地共轭标记到蛋白质上。
实施例5:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为生物素酰肼;
合成方法如下:
称取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢乙酸糠醇酯于烧瓶中,加入4mL 0.1M的HCl水溶液,室温搅拌3h。反应体系用碳酸氢钠固体中和至中性,加入0.4mL 500mM HEPES缓冲液(50mM,pH7.5)调节pH至7.5。向得到的混合物中加入129mg(0.5mmol,0.5eq)的生物素酰肼的DMSO(3mL)溶液,室温反应1h。旋蒸除去部分溶剂,剩余溶液过滤用反相HPLC分离,得棕色固体71mg(0.21mmol,产率42%)。分离条件:柱子:XB-C185-micron 10×250mm。溶剂A:水,B:乙腈。0-16min,B%:2%-30%。4mL/min,室温.RT=26min。1H NMR(400MHz,DMSO)δ10.28(br.,1H),7.41(d,3J=6.3Hz 1H),6.39(m,2H),5.77(s,1H),4.96(d,3J=39.4Hz,1H),4.31(d,3J=3.7Hz,1H),4.15(d,3J=3.7Hz 1H),3.11(m,2H),2.84(m,1H),2.58(m,1H),2.27(t,3J=6.8Hz,1H),2.06(m,1H),1.25-1.70(m,6H).13C NMR(101MHz,DMSO)δ171.5,166.8,162.7,143.8,136.9,123.5,96.9,61.0,59.2,55.3,40.4,32.8,28.0,27.9,24.9;ESI-Q-TOF:C55H21N4O3S,[MH]+cal.337.1334,found 337.1343。
5-亚甲基吡咯-2-酮类分子10对蛋白质中单个半胱氨酸的特异性标记反应:
将标记分子10(分子终浓度10mM)与组蛋白H4-R45C(终浓度50μM)混合于100mM NaCl,10mM HEPES(pH7.5)缓冲液中,37℃孵育2h。取样1μL注射到UPLC-ESI-Q-TOF系统中检测反应情况。图5表明,标记分子10能高效地共轭标记到蛋白质上。
实施例6:
一种N-取代-5-亚甲基-吡咯-2-酮类化合物的合成方法,所述N-取代-5-亚甲基-吡咯-2-酮类化合物的化学结构式为:
其中R为长链烷烃固相载体;
合成方法如下:
取202mg(1mmol,1eq)2,5-二甲氧基-2,5-二氢乙酸糠醇酯于烧杯中,加入4mL0.1M HCl,室温反应4h后,用碳酸氢钠固体中和反应体系,加入pH7.5的HEPES(终浓度50mM)缓冲液调节反应体系呈pH为7.5。直接在该反应体系中加入200mg LCCA-CPG(表面氨基的固相载体,氨基密度100μmol/g),室温下反应混合物在混合器上震荡4h。离心除去上清液,所得沉淀用去离子水洗涤两次,乙酸乙酯一次,最后用乙醚洗涤,自然晾干,得5MP-CPG。
5MP-CPG用于蛋白质在固相载体表面的固定:
选用的蛋白为大肠杆菌AHAS I酶的小亚基ilvN。ilvN含有两个半胱氨酸,可以通过与5MP-CPG表面5MP的迈克尔加成的反应而固定在CPG的表面。
取15mg 5MP-CPG,加入100μL ilvN(925μg/mL在HEPES缓冲液中,20mM,pH7.5,100mM NaCl),4℃下旋转混匀30min。通过bradford和12%SDS-PAGE分析显示上清中已无蛋白质。离心除去上清,所得沉淀中加入100μL的β-巯基乙醇(5mM水溶液)4℃下反应10min.以封闭未连接上ilvN的5MPs活性位点。离心所得沉淀用去离子水洗涤两次,得ilvN-CPG。
固相载体固定的蛋白ilvN-CPG用于蛋白复合体的亲和纯化,如图7所示:
大肠杆菌AHAS I酶的大亚基ilvB可以特异性地与ilvB非共价结合。因此利用ilvN-CPG可以将ilvB从细胞裂解液中选择性固定住,然后通过(I)高浓度的巯基乙醇或者(II)pH 9.5缓冲溶液处理,将ilvN-ilvB的复合体从固相载体上洗脱下来。具体操作过程如下:
(1)ilvB的固定。取15mg ilvN-CPG加入100μL细胞裂解液(内含ilvB蛋白),4℃下旋转混匀2h。移弃上清,沉淀用去离子水洗涤两次。得ilvB-ilvN-CPG。
(2)ilvN-ilvB复合体的洗脱。ilvN和ilvB从固相载体表面的洗脱分为四种方式:
(i)向15mg的ilvB-ilvN-CPG中加入100μL SDS loading buffer(50mM Tris,pH6.8,DTT 100mM,glycol 10%(v/v),SDS 2%(w/v),bromophenol 0.1%(w/v)),室温下孵育5min。离心,取上清10%SDS PAGE分析显示这种条件下ilvB被选择性洗脱下来,如图7所示。
(ii)向15mg的ilvB-ilvN-CPG中加入100μL SDS loading buffer(50mM Tris,pH 6.8,DTT 100mM,glycol 10%(v/v),SDS 2%(w/v),bromophenol 0.1%(w/v)),95℃孵育5min。离心,取上清用10%SDS-PAGE分析显示这种条件下ilvB和ilvN都被洗脱下来,如图7所示。
(iii)向15mg的ilvB-ilvN-CPG中加入100μL含700mM BME的HEPES缓冲液(20mM,pH 7.5,100mM NaCl)。4℃下旋转混匀2h。离心,取上清用10%SDS-PAGE分析显示ilvB和ilvN都被洗脱下来,酶活分析显示洗脱下来的复合体仍然具有AHAS全酶的活性,如图7所示。
(iv)向15mg的ilvB-ilvN–CPG加入100μL硼酸-氢氧化钠缓冲液(50mM,pH 9.5,100mM NaCl)。4℃下旋转混匀2h。离心,取上清用10%SDS-PAGE分析显示ilvB和ilvN都被洗脱下来,酶活分析显示洗脱下来的复合体仍然具有AHAS全酶的活性,如图7所示。
- 还没有人留言评论。精彩留言会获得点赞!