一种丙烯腈聚合物的绿色合成方法与流程
本发明属于高分子合成技术领域,具体涉及一种聚丙烯腈(PAN)的绿色合成方法。
背景技术:
作为碳纤维的前体,聚丙烯腈(PAN)具备刚性、抗张强度、化学容忍性等优点,在航空航天、机械制造、运动器材等行业一直有着举足轻重的地位。众所周知,具备高分子量、窄分子量分布、高立构规整度的聚丙烯腈是合成高质量的碳纤维材料的首要条件。然而,对于现有技术而言,丙烯腈的完全可控聚合仍然具有挑战性,主要是因为丙烯腈的高反应活性和聚丙烯腈不良溶解性。
1961年Shindo等人首先报道了以PAN作为基质的碳纤维材料的成功制备。随后,越来越多的碳纤维材料前体(PAN)通过本体、溶液、悬浮聚合等方法来制备。当然,副反应和苛刻的聚合条件一直是不可避免的因素,以至于这类碳纤维材料在机械性能的改善方面一直存在着局限。上世纪90年代,Nakano和Kunio首先报道了通过阴离子聚合方法成功制备出结构明确的聚丙烯腈,这为聚丙烯腈的结构表征提供了一个很好的开端。之后,包括原子转移自由基聚合(ATRP)、可逆加成-断裂链转移聚合(RAFT)、单电子转移活性自由基聚合(SET-LRP)在内的活性自由基聚合技术被运用于制备分子量可设计、分子量分布较窄、结构明确的聚丙烯腈的聚合物中。相比于其他的活性自由基聚合,RAFT技术具备高的官能团容忍性、无金属催化等有优点。然而,目前为止,也只有几个成功的案例,他们主要是利用2-苯基-2-丙基苯并二硫(CDB)、2-氰丙基-2-基苯并二硫(CPDB)、S,S-二苄基三硫代碳酸盐(DBTC)等作为链转移剂,但是仅仅通过热引发来制备聚丙烯腈聚合物。另外,60Co射线作为环境友好型能源资源,可以用来制备接枝、交联聚合物或者是用来修饰聚合物;但是这类的60Co射线辐照聚合难以控制。
技术实现要素:
针对上述情况,本发明公开了一种丙烯腈聚合物的绿色合成方法,在60Co射线辐照条件下,通过可逆加成-断裂链转移聚合方法(RAFT技术)实现室温下聚丙烯腈聚合物(PAN)的绿色可控合成;并且本发明的方法得到的聚丙烯腈分子量可控,分子量分布较窄。
为了达到上述目的,本发明公开了一种丙烯腈聚合物的绿色合成方法,包括如下步骤:将RAFT试剂、丙烯腈、有机溶剂加入反应器中;然后除氧处理;然后在60Co射线辐照下进行聚合反应1~10小时,得到丙烯腈聚合物;所述60Co射线辐照的剂量率为0.6~1.8 kGy/h。
上述合成方法得到丙烯腈聚合物,其中,RAFT试剂与丙烯腈摩尔比为100~1500∶1;得到的聚合物分子量适中、分子量分布比较窄。
上述技术方案中,用惰性气体进行除氧处理。
上述技术方案中,60Co射线为60Co-γ射线、60Co-β射线中任意一种,优选60Co-γ射线。
本发明还公开了一种丙烯腈聚合物的绿色合成方法,包括如下步骤:
(1)将RAFT试剂、丙烯腈、有机溶剂加入反应器中;然后除氧处理;然后在60Co射线辐照下进行聚合反应1~10小时,得到聚丙烯腈;所述60Co射线辐照的剂量率为0.6~1.8 kGy/h;
(2)将步骤(1)的聚丙烯腈、丙烯腈、有机溶剂加入反应器中;然后除氧处理;然后在60Co射线辐照下进行聚合反应1~10小时,得到丙烯腈聚合物;所述60Co射线辐照的剂量率为0.6~1.8 kGy/h。
上述技术方案中,步骤(1)中,RAFT试剂与丙烯腈摩尔比为100~1500∶1;制备的聚合物分子量适中、分子量分布比较窄,更有利于后期的扩链反应。
上述技术方案中,步骤(2)中,聚丙烯腈、丙烯腈、有机溶剂的质量比为(1~2)∶(40~100)∶(100~200);用惰性气体进行除氧处理;60Co射线为60Co-γ射线、60Co-β射线中任意一种,优选60Co-γ射线。
优选的,步骤(2)中,聚丙烯腈、丙烯腈、有机溶剂的质量比为(15~16)∶(400~420)∶(1300~1350)。
本发明中,RAFT试剂的制备方法为,将溴萘化合物、镁条、碘和反应溶剂加入到反应容器中,反应0.5~2小时,然后控制温度30~70℃条件下反应2~5小时;然后加入二硫化碳(CS2),继续反应;反应结束后,反应溶液倒入水中,加入酸化试剂进行酸化,然后利用萃取溶剂进行萃取,再用饱和食盐水洗,然后进行干燥浓缩,得到第一粗产物;第一粗产物用乙酸乙酯溶解,与二甲亚砜(DMSO)和少量碘反应过夜,通气条件下加入自由基引发剂和苯,在40~80℃条件下反应10~20小时,浓缩后得到第二粗产物,通过两次柱层析得到纯产物。
上述技术方案中,溴萘化合物选自1-溴萘、2-溴萘、2,6-二溴萘、2,8-二溴萘中的任意一种,优选1-溴萘;反应溶剂选自四氢呋喃、二氯甲烷、丙酮、正己烷中任意一种,优选四氢呋喃(THF);酸化试剂选自盐酸、硫酸、硝酸、醋酸中的任意一种,优选盐酸;自由基引发剂选自偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁酸二甲酯中的任意一种,优选偶氮二异丁腈(AIBN)。
本发明中,首先制备聚丙烯腈,然后作为大分子链转移剂进一步与丙烯腈进行聚合物反应,得到多种结构、分子量可控的丙烯腈聚合物。优选步骤(1)的聚丙烯腈的分子量Mn,GPC为1300~3900 g/mol,优选2000 g/mol;所限定的聚合物分子量适中、分子量分布比较窄,更有利于后期的扩链反应。
本发明中,有机溶剂选自二甲亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)、碳酸亚乙酯(EC)中任意一种,优选碳酸亚乙酯(EC)。EC对丙烯腈的溶解性更有优势,在聚合过程中可控性好,主要体现在分子量及其分子量分布上。
本发明中,利用惰性气体除氧,惰性气体选自氮气、氦气、氖气中的任意一种,优选氮气。氮气属于常规惰性气体,在同样的使用用途下,从经济角度思考,氮气价格更加便宜。
进一步的,上述方法中每一步反应完成之后都可以进行纯化步骤,以便得到纯度更高的产物,所述纯化步骤包括色谱法、溶解/沉淀分离法、过滤法等。
由于上述技术方案的实施,本发明与现有技术相比具有下列优点:
(1)本发明首次在链转移剂下,通过60Co射线辐照条件下,引发丙烯腈的辐照诱导活性自由基聚合,成功实现了丙烯腈的绿色合成,特别是本发明的方法只有三种组分,RAFT试剂、单体和溶剂,少于现有制备方法需要的组分,既避免了复杂副反应的发生,又解决了现有技术原料复杂、不环保的问题;
(2)本发明结合了活性自由基聚合方法(RAFT技术)和60Co-γ射线辐照方法,无需加热,在室温条件下实现了丙烯腈的活性可控聚合,与传统的热引发聚合相比,在工业上更具优势,为实现节约能源消耗、降低生产成本的提供了一个新的方向;
(3)本发明在聚合过程中,无金属催化剂使用,避免了金属污染问题,为制备以PAN为基质的碳纤维材料提供了一种新的绿色合成方法;
(4)由于丙烯腈有强极性基团的存在,这特殊基团的存在使得丙烯腈体现出高的反应活性,很难实现可控聚合,60Co射线辐照聚合一般情况下也很难实现可控聚合,而本发明结合了活性自由基聚合方法(RAFT技术)和60Co射线辐照聚合,得到的PAN的分子量可控、分子量分布也较窄,却实现了丙烯腈的活性可控聚合。
附图说明
图1为实施例2中部分聚丙烯腈的GPC流出曲线图;
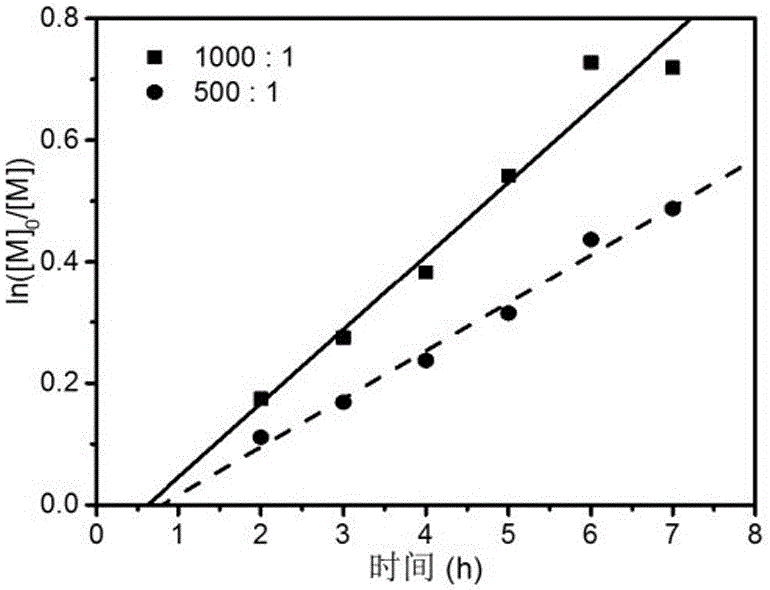
图2为相同辐照剂量率、不同比例下辐照聚合的动力学曲线图;
图3为不同比例下辐照聚合过程中Mn,GPC、Mw/Mn与单体转化率的演变图;
图4为相同比例、不同辐照剂量率下辐照聚合的动力学曲线图;
图5为不同辐照剂量率下辐照聚合过程中Mn,GPC、Mw/Mn与单体转化率的演变图;
图6为聚丙烯腈的核磁谱图;
图7为聚丙烯腈的质谱图;
图8为聚丙烯腈作为RAFT试剂引发丙烯腈、苯乙烯单体聚合前后的GPC流出曲线图;
图9为聚丙烯腈作为RAFT试剂引发丙烯腈、苯乙烯单体聚合前后的核磁谱图。
具体实施方式
本发明在活性自由基聚合方法(RAFT技术)的基础上,使用了60Co射线这一环境友好型能源资源,协同作用诱导丙烯腈的室温下活性/可控聚合,为制备以PAN为基质的碳纤维这类材料开拓了一种新的合成方法。下面将结合具体的实施例和附图对本发明做出进一步的描述。
化学试剂:
1-溴萘,化学纯,国药集团化学试剂有限公司;
镁,2N, 国药集团化学试剂有限公司;
碘,99.8%,江苏强盛功能化学股份有限公司;
四氢呋喃,99.5%,南京化学试剂有限公司;
二硫化碳(CS2),化学纯,国药集团化学试剂有限公司;
盐酸,12M, 江苏强盛功能化学股份有限公司;
二氯甲烷,99.5%,江苏强盛功能化学股份有限公司;
无水硫酸钠,98%,国药集团化学试剂有限公司;
乙酸乙酯,99.5%,江苏强盛功能化学股份有限公司;
二甲亚砜(DMSO),99.5%,江苏强盛功能化学股份有限公司;
偶氮二异丁腈(AIBN),化学纯,上海试剂四厂,使用前重结晶两次;
苯,99.5%,国药集团化学试剂有限公司;
石油醚,分析纯,江苏强盛功能化学股份有限公司
丙烯腈(AN),分析纯,使用前用短中性氧化铝柱纯化;
碳酸亚乙酯(EC),分析纯,国药集团化学试剂有限公司;
N,N-二甲基甲酰胺(DMF),99.5%,江苏强盛功能化学股份有限公司;
甲醇,分析纯,江苏强盛功能化学股份有限公司;
测试仪器及条件:
凝胶渗透色谱(GPC):分子量和分子量分布使用带有TOSOH TSKgel SuperHM-M的凝胶渗透色谱仪,属于自动进样式,聚苯乙烯作为标样计算聚合物分子量,N,N-二甲基甲酰胺(DMF)作为流动相,流速为0.65 mL/min,温度为40℃;
核磁共振氢谱(1H-NMR):使用Bruker 300MHz核磁仪,以CDCl3为溶剂,TMS为内标,室温下测定;
基质辅助激光解析电离飞行时间质谱(MALDI-TOF):使用Bruker Ultraflex-III MS spectrometer质谱仪,以DCTB为基质。
实施例1:RAFT试剂的合成
将1-溴萘 (22.5 g, 0.11 mol) 和四氢呋喃 (90 mL)缓慢地加入带有镁条 (2.88 g, 0.12 mol)和少量碘的250 mL三颈烧瓶中,所用时间约1小时,在40~50℃条件下反应3小时。室温条件下,将二硫化碳 (8.36 g, 0.11 mol)加入反应体系中,然后继续反应8小时。反应溶液倒入水中,并用2 mol/L的盐酸水溶液酸化,之后选用二氯甲烷 (40 mL×3)萃取。有机层用饱和食盐水洗,无水硫酸钠干燥。旋蒸除去溶剂之后,粗产品用50 mL乙酸乙酯溶解,与2.53 g DMSO、少量碘反应过夜。在通气条件下,向体系中加入AIBN (10.8 g, 0.066 mol) 和 40 mL 苯,并在70 ℃条件下反应15小时。旋蒸除去溶剂后,得到粗产物13.01 g。粗产物用硅胶柱柱层析来纯化,得到RAFT试剂CPDN;淋洗剂为石油醚:乙酸乙酯=10:1。1H NMR (CDCl3, 300 MHz), δ ppm: 8.10 (m, 1H), 7.85 (m, 2H), 7.50 (m, 2H), 7.43 (m, 2H), 1.96 (s, 6H)。
实施例2:一种丙烯腈聚合物的绿色合成方法
将实施例1得到的RAFT试剂、丙烯腈(AN)、溶剂碳酸亚乙酯(EC,2.64g)加入到5 mL的安焙瓶中,用氮气鼓泡法除氧,在60Co-γ射线辐照下发生聚合反应,室温反应丙烯腈聚合物。停止反应,得到聚丙烯腈聚合物。表1为不同条件下丙烯腈的辐照诱导自由基聚合结果。
表1不同条件下丙烯腈的辐照诱导自由基聚合结果
GPC测试中聚苯乙烯作为标样;称重法计算转化率;理论分子量计算方程为:
。
表1展示出一些不同条件下丙烯腈的辐照诱导自由基聚合结果,整体上可以看出聚合物分子量分布相对较窄,说明本发明基于活性自由基聚合(RAFT技术)基础上,再结合60Co射线辐照聚合,可以实现聚合物分子量分子布窄的目标。
图1为聚丙烯腈的GPC流出曲线图,相同的辐照剂量率、相同的反应时间,单体与RAFT试剂的比例越大,聚合速度越快,聚合物分子量越大,表现出来的GPC保留时间值越小。
图2为同一辐照剂量率、不同比例下辐照聚合的动力学曲线图;从图中可以明显看出,Ln([M]0/[M])与聚合时间呈一级线性关系,属于可控聚合,而单体与RAFT试剂的比例越大,聚合速度越快,相同时间的Ln([M]0/[M])值越大,这说明可以通过控制单体与RAFT试剂的比例或者反应时间的长短可以实现聚合物分子量的可设计性。
图3为不同比例下辐照聚合过程中Mn,GPC、Mw/Mn与单体转化率的演变图;图4为相同比例、不同辐照剂量率下辐照聚合的动力学曲线图;从图中可以明显看出,Ln([M]0/[M])与聚合时间呈一级线性关系,属于可控聚合,而辐照剂量率越大,聚合速度越快,相同时间的Ln([M]0/[M])值越大,这说明通过辐照剂量率的改变也可以实现聚合物分子量的可设计性。
图5为不同辐照剂量率下辐照聚合过程中Mn,GPC、Mw/Mn与单体转化率的演变图;图6为聚丙烯腈的核磁谱图;图7为聚丙烯腈的质谱图;说明本发明在活性自由基聚合方法、60Co射线协同作用下,诱导丙烯腈的室温下活性/可控聚合,为制备以PAN为基质的碳纤维这类材料开拓了一种新的合成方法。
实施例3:一种丙烯腈聚合物的绿色合成方法
将15.26 mg的PAN (表 1中1c)、0.41 g丙烯腈(AN)、1.32 g碳酸亚乙酯(EC)加入到5 mL的安焙瓶中,用氮气鼓泡法除氧,在60Co-γ射线辐照下发生聚合反应,反应时间为4小时。然后反应溶液用DMF(2 mL)溶解,然后用200 mL甲醇溶解;抽滤,得到的聚合物用真空干燥箱30℃干燥24小时,得到123.0 mg丙烯腈聚合物。
图8为聚丙烯腈作为RAFT试剂引发丙烯腈单体聚合前后的GPC流出曲线图,图9为聚丙烯腈作为RAFT试剂引发丙烯腈单体聚合前后的核磁谱图;从图中可以看出,聚丙烯腈作为RAFT试剂引发丙烯腈单体聚合前后分子量发生了变化,即分子量有所增长,说明聚合物末端依然可引发同类单体聚合,符合活性聚合特征,证明辐照诱导丙烯腈聚合属于活性聚合。
实施例4:一种丙烯腈聚合物的绿色合成方法
将15.2 mg的PAN (表 1中2c)、0.4 g丙烯腈(AN)、1.3 g碳酸亚乙酯(EC)加入到5 mL的安焙瓶中,用氮气鼓泡法除氧,在60Co-γ射线辐照下发生聚合反应,反应时间为2小时。然后反应溶液用DMF(2 mL)溶解,然后用200 mL甲醇溶解;抽滤,得到的聚合物用真空干燥箱30℃干燥24小时,得到118.8 mg丙烯腈聚合物,GPC分子量为48880g/mol,分子量分布为1.29。
实施例5:一种丙烯腈聚合物的绿色合成方法
将15.26 mg的PAN (表1中3b)、0.41 g丙烯腈(AN)、1.35 g碳酸亚乙酯(EC)加入到5 mL的安焙瓶中,用氮气鼓泡法除氧,在60Co-γ射线辐照下发生聚合反应,反应时间为10小时。然后反应溶液用DMF(2 mL)溶解,然后用200 mL甲醇溶解;抽滤,得到的聚合物用真空干燥箱30℃干燥24小时,得到121.2 mg丙烯腈聚合物,GPC分子量为38980g/mol,分子量分布为1.28。
- 还没有人留言评论。精彩留言会获得点赞!