一种含稠环芳烃结构多烷基取代的二胺化合物及其制备方法和应用与流程
本发明属于材料科学技术领域,更具体地,涉及一种含稠环芳烃结构多烷基取代的二胺化合物及其制备方法和应用。
背景技术:
聚酰亚胺是一类含有酰亚胺环结构的高分子材料,拥有优良的耐热性、化学稳定性、机械性能和介电性能等性能,现在已经被广泛应用于航天航空、电气、和汽车等许多领域。但是传统聚酰亚胺由于其分子链的高度共轭而导致在分子间和分子内形成电荷转移络合物(charge transfer complexs,CTC),使得聚酰亚胺溶解性差,颜色深,而限制了其在微电子以及光电子等高技术领域应用。因此,研发可溶和无色聚酰亚胺是聚酰亚胺材料的一个重要研究方向。
从结构上调控聚酰亚胺的溶解性和光学性能的方法主要集中于设计合成含非共轭基团的二胺或二酐。目前,非共轭基团包括柔性醚键(Macromolecules,2006,39,7555–7560)、大位阻侧基(Chem.Mater.,2001,13,2472–2475)、螺旋结构、含氟基团(J.Polym.Sci.Part A:Polym.Chem.,2013,51,4413–4422)和脂肪单元(Macromol.Rapid.Commun.,2000,21,1166–1170;J.Polym.Sci.Part A:Polym.Chem.,2009,47,3309-3317)等。但这些非共轭基团的引入都有一定局限性,譬如引入醚键和脂肪单元往往会降低材料的热稳定性和机械性能,引入三氟甲基会影响聚合物的尺寸稳定性。
因此,近来人们更关注把不同基团同时引入到同一种单体中,以期在改善聚酰亚胺的溶解性和光学透明性的同时,不影响材料的其它性质。其中,含芳香侧基的多烷基取代的二胺化合物由于既有烷基的位阻效应又兼具芳香基的稳定性,而备受青睐。中国专利CN1206203A和CN1970527A报道了一类含芳香侧基的多烷基取代的二胺单体。这些专利报道的多烷基取代的二胺单体的侧基都只是苯基或氟化苯类衍生物,同时,烷基取代位置单一。
技术实现要素:
本发明的目的在于根据现有技术中的不足,提供了一种含稠环芳烃结构多烷基取代的二胺化合物。
本发明的另一目的在于提供上述二胺化合物的制备方法。
本发明的再一目的在于提供上述二胺化合物的应用。
本发明所合成的含稠环芳香结构多烷基取代的二胺化合物,由于稠环芳烃具有更大位阻效应,由该类二胺单体所制备的聚合物分子链相互作用更小,自由体积更大,而使材料具有优异的溶解性和无色透明性。
本发明的目的通过以下技术方案实现:
本发明提供了一种含稠环芳烃结构多烷基取代的二胺化合物,结构式如式(I)或式(II)所示:
其中,X为氢或烷基;Y为氢、苯基、取代苯基、稠环芳香基、羟基或卤基。
优选地,X为氢或C1~5烷基。
最优选地,X为氢、甲基或异丙基。
优选地,Y为稠环芳香基。
优选地,X为氢或C1~3烷基。
优选地,Y选自如下基团中的一种:
最优选地,Y为
本发明同时提供所述的二胺化合物的制备方法,包括将醛类化合物Y-CHO与取代苯胺衍生物在质子酸的催化下反应得到;
所述取代苯胺衍生物的结构式如式(III)或式(IV)所示:
其中,X为氢或烷基;Y为氢、苯基、取代苯基、稠环芳香基、羟基或卤基。
优选地,所述质子酸为盐酸、硫酸、三氟乙酸、三氟甲磺酸、对甲苯磺酸中的一种或多种。
优选地,所述反应温度为80~160℃。
优选地,所述醛类化合物Y-CHO与取代苯胺衍生物的反应摩尔比为1:(2~8)。
与现有技术相比,本发明具有以下优点及有益效果:
1、本发明所提供的含稠环芳烃结构多烷基取代的二胺化合物,在分子结构上稠环芳烃位阻大,烷基数目多,有利于结构调控,以其为单体制备的聚酰亚胺分子链的结构扭曲,堆砌紧密度小,分子链内和链间CTC效应受到抑制。同时,由于稠环芳烃结构稳定性高,有利于保持聚合物的热稳定性。
2、本发明的二胺化合物由取代甲醛与多烷基苯胺衍生物反应一步而制得,合成简单,纯化容易,原料易得,适合工业化生产。
3、本发明的含稠环芳烃结构多烷基取代的二胺单体在制备可溶、无色透明的聚酰亚胺中具有很大的潜力,同时也适用于合成聚酰胺和聚酰胺酰亚胺高性能材料。
附图说明
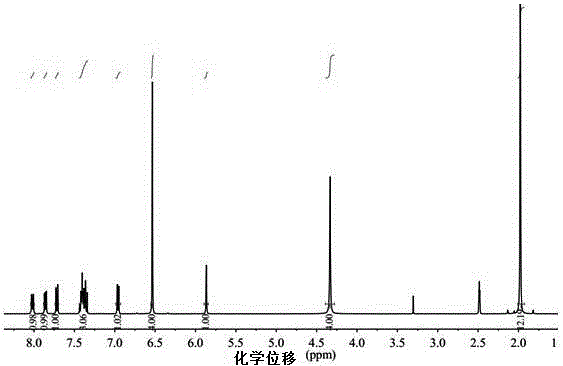
图1.本实施例1由苯胺和1-萘甲醛合成的二胺化合物的1H NMR谱图。
图2.本实施例2由2、6-二甲基苯胺和1-萘甲醛合成的二胺化合物的1HNMR谱图。
图3.本实施例3由2、6-二异丙基苯胺和1-萘甲醛合成的二胺化合物的1H NMR谱图。
图4.本实施例4由2、4-二甲基苯胺和1-萘甲醛合成的二胺化合物的1H NMR谱图。
具体实施方式
以下结合具体实施例和附图来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,本发明所用试剂和材料均为市购。
实施例1
本实施例合成的二胺结构式如下所示:
上述二胺的合成步骤如下:将16.63g苯胺置于100mL三口瓶中,搅拌,通氮气置换瓶内空气,加热到130℃后,用滴液漏斗将7.87g 1-萘甲醛和4.2mL的浓盐酸混合溶液滴入反应体系中,保持该温度反应18h,反应体系变稠,为深棕色。反应结束后体系降温至60℃,在搅拌下滴加10g 20wt%的氢氧化钠水溶液后,滴入100mL甲醇中,析出固体。经过滤,甲醇和水多次洗涤固体后,用甲醇重结晶,析出棕黄色晶体,过滤收集晶体,在60℃下真空干燥12h,得到6.93g产物,产率为24%。
产物熔点:219~220℃。
红外光谱(KBr,cm-1):3436,3416,3337,3212,3025,1619,1509,1271,1176,830,800,572,507。
核磁共振氢谱(400MHz,DMSO-d6,ppm),δ=8.04(d,J=8.0Hz,1H),7.89–7.85(m,1H),7.73(d,J=8.2Hz,1H),7.45–7.34(m,3H),6.93(d,J=7.1Hz,1H),6.74(t,J=7.9Hz,4H),6.48(t,J=8.3Hz,4H),5.94(s,1H),4.86(s,4H)。
元素分析:found:C.84.86%,H.5.903%,N.8.81%;Calc.:C.85.15%,H.8.21%,N.8.63%。
实施例2
本实施例合成的二胺结构式如下所示:
上述二胺合成步骤如下:15.59g 2、6-二甲基苯胺置于100mL三口瓶中,,搅拌,通氮气置换瓶内空气,加热到80℃。4.29g 1-萘甲醛与2.5mL浓盐酸充分混合使其活化,并在1.5h内把活化1-萘甲醛溶液滴入到反应体系中。然后加热到120℃,回流反应10h。随着反应的进行,反应体系由棕色溶液变为蓝色,最终变为蓝色粘稠液体。体系降到60℃后,加入7.5g 20wt%的氢氧化钠水溶液。再降温到室温,在搅拌下,把反应体系加入到200mL甲醇中,析出淡黄色固体。经过滤,甲醇和水多次洗涤固体后,用甲醇重结晶,析出浅灰色晶体,过滤收集晶体,在60℃下真空干燥12h后,得到3.60g产物,产率为36%。
产物熔点:214~215℃。
红外光谱(KBr,cm-1):3448,3396,2972,2924,1623,1956,1490,1438,1307,1145,883,788,736。
核磁共振氢谱(400MHz,DMSO-d6,ppm),δ=8.02(d,J=7.7Hz,1H),7.88–7.84(m,1H),7.72(d,J=8.2Hz,1H),7.44–7.33(m,3H),6.96(d,J=7.1Hz,1H),6.53(s,4H),5.87(s,1H),4.33(s,4H),1.98(s,12H)。
元素分析:found:C.85.06%,H.7.469%,N.7.47%;Calc.:C.85.22%,H.7.42%,N.7.36%。
实施例3
本实施例合成的二胺结构式如下所示:
上述二胺合成步骤如下:依次将22.33g 2、6-二异丙基苯胺、15mL水、30mL乙二醇甲醚加于100mL三口瓶中,通氮气置换瓶内空气,加热到90℃。7.5mL浓硫酸缓慢滴入瓶内,获得无色透明溶液,再滴入10.40g 1-萘甲醛,升温至100℃,反应14h。反应体系中出现黄色沉淀物。反应结束后降温至室温,将反应体系加入800mL水中,调pH至9~10,搅拌4h,经过滤获得固体,用大量水反复洗涤固体。将固体溶入二氯甲烷,水萃取三遍,除去无机盐,旋蒸除去二氯甲烷,得到白色固体,然后用甲醇重结晶,析出白色晶体,过滤收集晶体,在60℃下真空干燥12h后,得到7.20g产物,产率为49%。
产物熔点:132~133℃。
红外光谱(KBr,cm-1):3460,3385,2959,2869,1622,1595,1465,1392,1353,1287,786。
核磁共振氢谱(400MHz,DMSO-d6,ppm),δ=8.24–8.17(m,1H),7.88–7.82(m,1H),7.70(d,J=8.2Hz,1H),7.41(dd,J=9.6,5.0Hz,2H),7.37(t,J=5.4Hz,1H),6.97(d,J=7.1Hz,1H),6.71(s,4H),5.99(d,J=8.7Hz,1H),4.35(s,4H),2.95(hept,J=6.6Hz,4H),1.02(dd,J=8.2,7.0Hz,24H)。
元素分析:found:C.85.43%,H.9.035%,N.5.57%;Calc.:C.85.31%,H.9.00%,N.5.69%。
实施例4
本实施例合成的二胺结构式如下所示:
上述二胺合成步骤如下:将15.61g 2、4-二甲基苯胺加于100mL三口瓶中,通氮气置换瓶内空气,加热到120℃。将4.17g 1-萘甲醛与2.5mL浓盐酸混合溶液滴入反应体系中,保持120℃反应15h后,升温到140℃反应1h,最终体系变为黄色粘稠液。体系降温到60℃后,加入7.5g 20%的氢氧化钠水溶液,然后降温到室温,在搅拌下,反应体系滴入200mL甲醇中,析出棕黄色的固体。经过滤,甲醇和水多次洗涤固体后,用甲醇重结晶,获得黄色固体,过滤收集固体,在60℃下真空干燥12h后,得到2.10g产物,产率为22%。
红外光谱(KBr,cm-1):3445,3416,3363,2865,2859,1642,1541,1521,1490,1439,1394,1255,866,796,776,675,560。
核磁共振氢谱(400MHz,DMSO-d6,ppm),δ=7.93(d,J=7.6Hz,1H),7.82(d,J=8.1Hz,2H),7.51–7.38(m,3H),6.94(d,J=7.1Hz,1H),6.72(s,2H),6.19(s,2H),6.03(s,1H),4.13(s,4H),2.10(s,6H),1.96(s,6H)。
元素分析:found:C.85.64%,H.7.14%,N.7.10%;Calc.:C.85.22%,H.7.42%,N.7.36%。
将本发明实施例提供的二胺化合物制备成聚酰亚胺,其中,以实施例1~3中提供的化合物为例,分别与反应生成相应的聚酰亚胺,其溶解性和光学透过性分别见表1和表2所示。
表1 不同二胺化合物合成的聚酰亚胺的溶解性
++:在室温下溶解.
+:在加热到100℃下溶解.
+-:在加热到100℃或沸点温度下部分溶解.
-:加热不溶解.溶解度为在10mg/mL的浓度下加热24小时后判定.
1/ODPA代表实施例1制备的二胺化合物与ODPA制备得到的聚酰亚胺材料,其它类推。
表2 不同二胺化合物合成的聚酰亚胺的光学透过性
其中,λ0%/nm代表透过率为0%的波长,T400nm/%代表在400nm波长下的透光率。
从表1和表2中来看,实施例3提供的材料的溶解性和透光率最佳,尤其适合作为制备功能性聚酰亚胺材料。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种烷基芳基取代噁二唑化合物的三组分合成方法与制造工艺
- N‑萘烷基取代的含氮杂环衍生物、制备方法及其抗肿瘤用途与制造工艺
- 一种环戊二烯基钠—四氢呋喃溶液生产制备装置的制造方法
- 一类噁唑并香豆素衍生物在农药方面的应用
- 一种2?烷基蒽醌的合成方法
- 5?氟?4?亚氨基?3?(烷基/取代烷基)?1?(芳基磺酰基)?3,4?二氢嘧啶?2(1h)?酮及其制备方法
- 5?氟?4?亚氨基?3?(烷基/取代烷基)?1?(芳基磺酰基)?3,4?二氢嘧啶?2(1h)?酮及其制备方法
- 一种基于氢核磁共振快速测定普瑞巴林原料药纯度的方法
- 包含乙烯、乙烯基酯和(甲基)丙烯酸酯的共聚物,其配制剂和作为原油的倾点下降剂、蜡 ...的制作方法
- 一种n-芳基磷代甲酰胺的制备方法