无机颗粒‑聚硅氧烷复合物、包含复合物的分散体和固体材料、以及制备方法与流程
本非临时申请在美国法典第35卷第119节(a)款下要求2015年12月8日于日本提交的第2015-239303号的专利申请的优先权,所述专利申请的全部内容通过引用并入本文。
技术领域
本发明涉及无机颗粒-聚硅氧烷复合物,包含所述复合物的分散体,包含所述复合物的固体材料和制备所述复合物的方法。更特别地,其涉及聚硅氧烷接枝至无机颗粒表面的无机颗粒形式的材料,其显示出有机硅的透明性和耐候性以及无机颗粒的功能。
发明背景
由于优异的透明度和耐候性,有机硅材料用于各种应用。最近努力通过向常规有机硅材料中引入具有某种功能如吸光性、耐热性、导电性或磁性的组分而将其转变成官能化的有机硅材料。例如,专利文献1公开了其中引入有UV吸收性有机分子的UV吸收性涂料组合物,专利文献2公开了负载有无机填料的耐热硅橡胶材料。
在官能化的有机硅材料的制备中,通常将待引入的官能化组分分成有机分子和无机颗粒。许多有机分子与有机硅材料完全相容,从而可获得高度透明的材料。然而,因为有机分子比有机硅材料耐候性差,在长期使用期间的功能退化和变色是不可避免的。另一方面,大多数无机颗粒显示出比有机硅材料更好的耐候性,并且因此使在长期使用期间的功能退化和变色的风险最小化。然而,因为无机颗粒在硅氧烷材料中相容差或不可分散,所以它们倾向于在引入后聚集,导致透明度和功能的实质损失。
无机颗粒在硅油中由于以下原因难于稳定分散:(1)因为硅油是非极性液体,所述典型地为亲水性的无机颗粒倾向于聚集,和(2)在粘性液体如硅油中,无机颗粒通过布朗运动的扩散运动弱并且因此分散受到抑制。因此,与其它有机油和树脂相比,难于制备无机颗粒稳定地分散在其中的透明的功能性有机硅材料。因此相信难于制备基于高分子量硅油和负载无机颗粒的透明硅油或橡胶材料。
改进无机颗粒在有机溶剂中的分散性的手段中,一种有效的途径是用具有大的排除体积的取代基改性颗粒表面,由此避免颗粒之间的直接接触。目前报道了对无机颗粒用硅烷在其表面上进行处理。由此经表面处理的无机颗粒具有在有机溶剂中的高分散性。发明人先前在专利文献3中提出通过使涂覆有氧化硅水合物的纳米颗粒状氧化钛的水分散体与硅烷偶联剂反应获得具有纳米量级颗粒尺寸的醇分散体。由该方法获得的纳米颗粒可完全分散在极性有机溶剂如醇和聚醚溶剂中,但是难于分散在硅油中。
专利文献4描述了聚硅氧烷接枝至其上的无机颗粒,其通过用砂磨机研磨和混合在甲苯溶剂中的在一端具有反应性基团的聚硅氧烷和无机颗粒来合成。将由此处理的无机颗粒引入有机硅包封剂。将聚硅氧烷接枝至无机颗粒的表面有效改进无机颗粒的分散性。然而,必须预先合成仅在一个端部精确官能化的聚硅氧烷化合物,这从工作效率的方面来看是不期望的。另外,因为在聚合物末端的反应性随着分子量的增加而降低,该技术难于将聚硅氧烷以高密度接枝至颗粒。对颗粒的分散性施加了诸多限制。此外,该技术要求机械单元操作如研磨以分散无机颗粒。研磨操作需要大量能量。可能使一旦聚集的颗粒在颗粒尺寸分布和颗粒形状方面较聚集之前的状态大大改变。所述技术因此在工业上是不利的。
缺少机械单元操作如研磨的方式公开于专利文献5中。通过使预先合成的聚硅氧烷在无机颗粒的甲醇分散体中反应而进行无机颗粒的表面处理。可用于该技术中的聚硅氧烷被限制在可溶于甲醇中的低分子量的那些。完全没有描述经表面处理的无机颗粒可分散于硅油中。
引用文献列表
专利文献1:JP 5353843
专利文献2:JP-A 2013-035890
专利文献3:JP-A 2015-034106
专利文献4:JP 5472543(WO 2013/133430)
专利文献5:WO 2013/094738
发明公开
考虑到
背景技术:
,发明人面对以下问题。为了生产具有分散于其中的无机颗粒的透明的功能性有机硅组合物,需要具有在硅油中的高分散性的无机颗粒状材料。为此,将高分子量的聚硅氧烷以高密度接枝至无机颗粒的表面是有效的。如专利文献4中所公开的接枝具有高分子量的单末端功能聚硅氧烷的技术难于将聚硅氧烷以高密度接枝至无机颗粒的表面,因为在聚合物末端的反应性差。此外,因为许多高分子量聚硅氧烷在极性溶剂中不溶,所以它们难于在保持分散的状态下与亲水性无机颗粒反应,同时。另一方面,当控制待反应的硅氧烷低聚物的分子量至其可溶于如专利文献5中所描述的极性溶剂的水平时,不能期望令人满意的分散性。可以预期通过机械研磨处理将无机颗粒分散在非极性溶剂中。该技术在工业上是不利的,因为研磨操作需要大量能量并且可能使一旦聚集的颗粒在颗粒尺寸分布和颗粒形状方面较聚集之前的状态大大改变。
从未提出过提供有用的结果并且有助于相关工业开发的同时解决矛盾的问题的技术。
本发明的目的在于提供无机颗粒-聚硅氧烷复合物,其可以分散在有机硅流体中以形成透明材料;制备所述复合物的方法;和具有无机颗粒的功能与有机硅流体的透明和机械性质二者的材料。
在寻求无机颗粒透明地分散于其中的功能性有机硅材料时,发明人已发现通过在无机颗粒在作为分散介质的极性有机溶剂中的分散体中进行环状硅氧烷的开环聚合,可以制备包含聚硅氧烷接枝至它们的表面的且可透明地分散在硅油中的无机颗粒的无机颗粒-聚硅氧烷复合物,而不需要机械处理如研磨。由此获得的无机颗粒-聚硅氧烷复合物可充分分散于高分子量硅油中。推测紧随开环之后,环状硅氧烷以低分子量低聚物的形式高密度地接枝至无机颗粒的表面。随着通过除去分散介质将体系浓缩,进行缩合,即高分子链生长。随着从体系中除去分散介质使得环境逐渐变成疏水性,被覆颗粒的聚硅氧烷逐渐从低聚状态转变成高分子量状态,表明颗粒变得更加疏水。因此,从颗粒分散于极性溶剂中的状态最终获得其中无机颗粒包封在有机硅组分内的无机颗粒-聚硅氧烷复合物。本发明使得在现有技术中被认为是困难的于常规反应器中不需要机械研磨处理用疏水性高分子量聚硅氧烷处理亲水性无机颗粒成为可能。
因此,本发明提供了可分散于硅油中的无机颗粒-聚硅氧烷复合物,包含所述复合物的分散体,包含所述复合物的固体材料,和制备所述复合物的方法,如下文所定义。
在一个方面,本发明提供了无机颗粒-聚硅氧烷复合物,其包含无机颗粒和至少部分接枝至所述颗粒表面的包含SiR2R3O2/2和SiR4R5R6O1/2单元的聚硅氧烷,所述无机颗粒具有通过动态光散射法测量的体积基准颗粒尺寸分布中的1至200nm的50%累积颗粒尺寸,所述聚硅氧烷任选地经由包含SiR1O3/2单元的硅氧烷涂覆层结合至所述无机颗粒表面,和由SiR1O3/2、SiR2R3O2/2和SiR4R5R6O1/2单元表示的硅氧烷成分的合计为20至20,000重量份,按每100重量份的所述无机颗粒计。其中,R1独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基、氨基、巯基、异氰酸酯或氟基团取代,R2、R3、R4和R5各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基、或氟基团取代,和R6为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基、具有至多50个硅原子的(聚)二甲基甲硅烷氧基、C1-C10烷氧基和羟基,它们可以被(甲基)丙烯酰基、环氧乙烷基、或氟基团取代。
在优选的实施方案中,无机颗粒包括至少一种选自以下的化合物:氧化铝、氧化铈、氧化钛、氧化锌、氧化铟锡、氧化锆、氧化锡、氧化铁和氧化硅。
在优选的实施方案中,无机颗粒为核/壳结构,其具有包括选自氧化铝、氧化铈、氧化钛、氧化锌、氧化铟锡、氧化锆、氧化锡和氧化铁的至少一种化合物的核和围绕所述核的氧化硅的壳。
在优选的实施方案中,在通过29Si-NMR光谱法分析时,计算归属于全部D单元(SiR2R3O2/2)的NMR信号的积分值(ΣD)与归属于全部M单元(SiR4R5R6O1/2)的NMR信号的积分值(ΣM),且ΣD/ΣM的比例为至少3。
在另一方面,本发明提供了包括在介质中的如上文定义的无机颗粒-聚硅氧烷复合物的分散体。
在优选的实施方案中,介质为选自戊烷、己烷、庚烷、壬烷、癸烷、苯、甲苯、邻二甲苯、间二甲苯、对二甲苯和聚硅氧烷化合物中的至少一种化合物。在更优选的实施方案中,所述聚硅氧烷化合物为选自以下的至少一种化合物:六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷和十二甲基环六硅氧烷。典型地,聚硅氧烷化合物为在25℃时具有50至100,000mm2/s的动态粘度的聚二甲基硅氧烷。
在另一个方面,本发明提供了包括如上文所定义的无机颗粒-聚硅氧烷复合物的固体材料。所述固体材料还可以包括粘结剂,所述粘结剂为有机硅树脂或硅橡胶。
在又一方面,本发明提供了用于制备如上文定义的无机颗粒-聚硅氧烷复合物的方法,包括如下步骤:
(a)提供无机颗粒在极性有机溶剂中的分散体,所述无机颗粒具有通过动态光散射法测量的1至200nm的体积基准50%累积颗粒尺寸,和
(b)混合所述分散体与至少一种具有通式(1)的环状硅氧烷,并使混合物中的所述环状硅氧烷进行开环聚合。
其中,R7、R8、R9、R10、R11和R12各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基、或氟基团取代,n为0至10的整数。
在优选的实施方案中,步骤(a)包括将具有1至200nm的体积基准50%累积颗粒尺寸的无机颗粒用硅烷化合物和/或其(部分)水解缩合物表面处理并将所述颗粒分散在极性有机溶剂中。
在优选的实施方案中,在步骤(a)中的极性有机溶剂为选自以下的至少一种溶剂:甲醇、乙醇、异丙醇和正丙醇。
在优选的实施方案中,步骤(b)在酸催化剂或碱催化剂存在下进行。
在优选的实施方案中,所述环状硅氧烷为选自以下的至少一种化合物:六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环六硅氧烷和2,4,6,8-四甲基环四硅氧烷。
在优选的实施方案中,步骤(b)包括在基于环状硅氧烷计至多1摩尔当量的量的作为聚合物扩链组分的硅烷化合物和/或其水解缩合物存在下的环状硅氧烷的开环聚合,所述硅烷化合物具有通式(2):
R13R14Si(OR15)2 (2)
其中,R13和R14各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酸酰基、环氧乙烷基或氟基团取代,R15为氢或具有至多6个碳原子的烷基。
在优选的实施方案中,步骤(b)包括在按100重量份所述环状硅氧烷计至多100重量份的量的作为聚合物链终端组分的硅化合物存在下的环状硅氧烷的开环聚合,所述硅化合物具有通式(3)或(4):
R16R17R18Si(X) (3)
R16R17R18SiOSiR16R17R18 (4)
其中,R16、R17和R18各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代,X为选自烷氧基、烯丙基、乙酰氧基、烯醇和氯的基团。
所述方法可以还包括步骤(c):在步骤(b)之后除去极性有机溶剂。
所述方法可以还包括在步骤(b)之后的步骤(d):添加式(1)的环状硅氧烷、式(2)的聚合物扩链组分和/或式(3)或(4)的聚合物链终端组分,并再次进行反应。
发明的有益效果
可以将本发明的无机颗粒-聚硅氧烷复合物分散在硅油和其他介质中,以形成透明材料。
附图简述
图1为显示实施例1和10中的聚硅氧烷接枝的无机氧化物(无机氧化物颗粒-聚硅氧烷复合物)TP-1和TP-6的1H-NMR光谱的图。
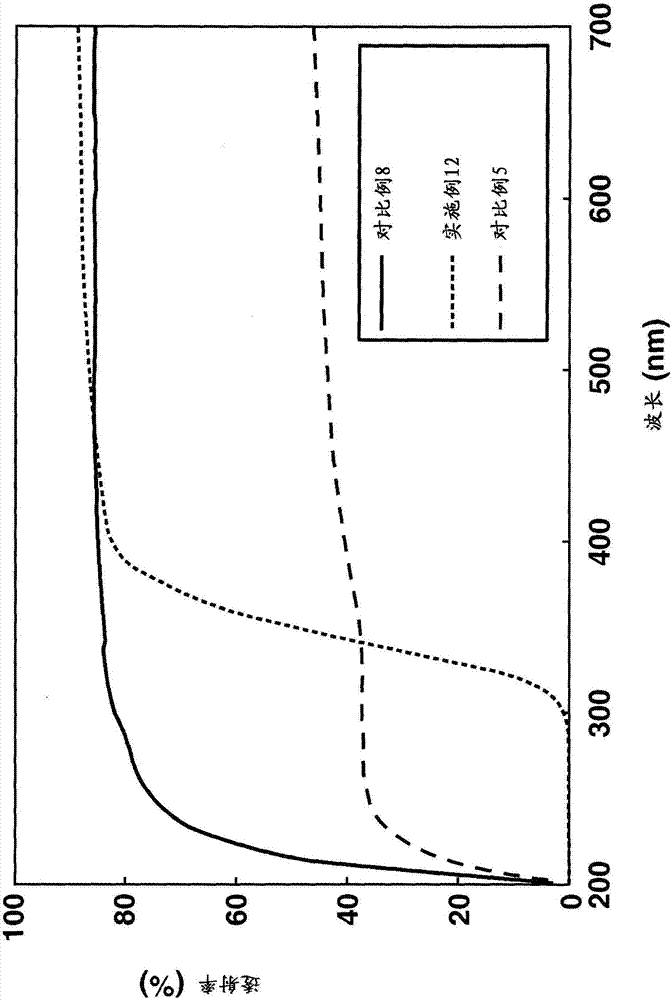
图2为显示实施例12和对比例5和8中的硅橡胶的UV-Vis透射光谱的图。
图3为显示实施例13和对比例6和8中的硅橡胶的UV-Vis透射光谱的图。
图4为显示实施例14和对比例7和8中的硅橡胶的UV-Vis透射光谱的图。
优选实施方案的描述
如本文中所使用的标记法(Cn-Cm)表示每个基团包含n至m个碳原子的基团。Me表示甲基。缩写:NMR为核磁共振光谱,GPC为凝胶渗透色谱,DOP为聚合度,TGA为热重分析。有时将无机颗粒-聚硅氧烷复合物简称为“复合物”。
现在详细地描述无机颗粒-聚硅氧烷复合物,包含所述复合物的分散体和固体材料,和制备所述复合物的方法。
[无机颗粒-聚硅氧烷复合物]
将无机颗粒-聚硅氧烷复合物定义为包含无机颗粒和至少部分接枝至所述颗粒表面的包含SiR2R3O2/2和SiR4R5R6O1/2单元的聚硅氧烷。所述无机颗粒具有通过动态光散射法测量的体积基准颗粒尺寸分布中的1至200nm的50%累积颗粒尺寸。所述聚硅氧烷可以经由包含SiR1O3/2单元的硅氧烷层结合至所述无机颗粒表面。由SiR1O3/2、SiR2R3O2/2和SiR4R5R6O1/2单元表示的硅氧烷成分的合计为20至20,000重量份,按每100重量份的所述无机颗粒计。
在此,R1独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基、氨基、巯基、异氰酸酯或氟基团取代。R2、R3、R4和R5各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代。R6为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基、具有至多50个硅原子的(聚)二甲基甲硅烷氧基、C1-C10烷氧基和羟基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代。
接枝的聚硅氧烷基本上由SiR2R3O2/2和SiR4R5R6O1/2单元组成,但可以任选地包含SiO2和R2SiO3/2单元。硅氧烷涂覆层基本上由SiR1O3/2单元组成,但可以任选地包含D单元和/或Q单元。
在此,除非另有指明,Q单元是指SiO4/2结构的单元,T单元是指SiR1O3/2结构的单元,D单元是指SiR2R3O2/2结构的单元,和M单元是指SiR4R5R6O1/2结构的单元。另外,除非另有指明,M单元可以具有至少一个在硅上取代的氧原子,只要它们是接枝结构部分中的聚硅氧烷末端成分。例如,R6可以为烷氧基或羟基。
聚硅氧烷组分
在R1、R2、R3、R4、R5和R6中,所述C1-C20烷基优选为具有1至6个碳原子的那些并且包括甲基、乙基、丙基和丁基。尤其最优选的是甲基。所述C2-C20烯基优选为具有2至6个碳原子的那些,更优选乙烯基和烯丙基。所述C1-C10烷氧基优选为具有1至6个碳原子的那些,并且包括甲氧基、乙氧基、丙氧基和丁氧基。尤其最优选的是甲氧基。所述C6-C20芳基优选为具有6至10个碳原子的那些,并且最优选的是苯基、甲苯基和二甲苯基。具有至多50个硅原子的(聚)二甲基甲硅烷氧基优选为具有1至50个硅原子、更优选1至30个硅原子的那些。
当R1、R2、R3、R4、R5和R6为甲基时,可以获得具有储存稳定性和耐热性的无机颗粒复合物。当R1、R2、R3、R4、R5和R6中的一些为苯基时,可以获得对苯基有机硅材料具有高亲和性的无机颗粒复合物。当R1、R2、R3、R4、R5和R6中的至少一个为氢、乙烯基或烯丙基时,可以获得的有利之处在于,当无机颗粒复合物与有机硅组合物的混合物固化时,可以获得其中无机颗粒交联至有机硅组合物的韧性材料,因为所述相关基团经历与有机硅组合物中的氢有机硅或乙烯基有机硅的氢化硅烷化反应。当R6为羟基或烷氧基时,可以通过与各种环状硅氧烷或烷氧基硅烷反应将有机硅链延长或末端改性。
无机颗粒-聚硅氧烷复合物中的T单元的合计优选为0至100重量份,更优选10至70重量份,且甚至更优选10至50重量份,按每100重量份的无机颗粒计。如果T单元的合计超过100重量份,则许多T单元结合至颗粒表面,而将颗粒表面上的羟基封端,这可能阻止接枝结构部分有效地结合至无机颗粒。
无机颗粒-聚硅氧烷复合物中的D单元的合计优选为10至10,000重量份,更优选50至1,000重量份,且甚至更优选100至500重量份,按每100重量份的无机颗粒计。如果D单元的合计小于10重量份,则聚硅氧烷被覆量可能过低而不能避免无机颗粒之间的直接接触,导致在非极性溶剂中的差的分散性。如果D单元的合计超过10,000重量份,则无机颗粒在无机颗粒-聚硅氧烷复合物中的重量份额相对低,无机颗粒可能无法发挥它们的功能。
优选地,无机颗粒-聚硅氧烷复合物满足以下关系:0<(M单元数)<(D单元数)。如果M单元数大于或等于D单元数(这意味着存在更多的聚合物链末端组分),则接枝的聚硅氧烷可能具有过低的分子量而不能避免无机颗粒之间的直接接触,导致在非极性溶剂中的差的分散性。M单元数与D单元数的比例优选为至多0.5/1,更优选至多0.2/1。下限优选为至少0.001/1。
无机颗粒-聚硅氧烷复合物应当以20至20,000重量份,优选50至10,000重量份的量包含聚硅氧烷组分,按每100重量份的无机颗粒计。少于20重量份的聚硅氧烷组分可能无法避免无机颗粒之间的直接接触,导致在非极性溶剂中的差的分散性。大于20,000重量份的聚硅氧烷组分意味着无机颗粒在复合物中的相对份额低,无机颗粒可能无法发挥它们的功能。
所引入的聚硅氧烷的量可以通过将在热重分析(TGA)之后的残余物的重量测定为无机颗粒的重量并将重量损失测定为聚硅氧烷组分的重量来计算。对于TGA,可以使用Rigaku Corporation的仪器Thermo Plus。分析优选在惰性气氛中进行以防止与氧反应。
接枝结构部分指定为通过无机颗粒的表面处理而接枝至其表面的聚硅氧烷组分。虽然二氧化硅颗粒包含SiO2单元(Q单元)或核/壳型颗粒在其壳中包含Q单元硅,但是在颗粒本身中的这些硅氧烷成分并不包括在接枝结构部分中。另一方面,当将无机颗粒用在一端用三甲氧基甲硅烷氧基封端(即在一端具有反应性Q单元)的聚硅氧烷(例如Shin-Etsu Chemical Co.,Ltd.的X-24-9822)表面处理时,结合至颗粒表面的来自X-24-9822的Q单元包括在接枝结构部分中。
即,接枝结构部分是指结合至颗粒的表面上的反应性基团如OH和甲氧基的线形聚硅氧烷结构部分。
无机颗粒
无机颗粒包含形成核的元素,所述元素优选选自第13族元素、第14族元素(排除碳)、第一系列过渡元素、第二系列过渡元素、第三系列过渡元素和镧系元素。第13族元素中,更优选的是铝的氧化物、硼的氧化物和铟的氧化物。第14族元素(排除碳)中,更优选的是金属硅颗粒与锡的氧化物和硅的氧化物。第一系列过渡元素中,更优选的是钛的氧化物、锰的氧化物和锌的氧化物。通常将这些氧化物用作用于吸收特定波长的光的试剂。第二系列过渡元素中,更优选的是银颗粒与钇的氧化物和锆的氧化物。通常将这些氧化物用作用于吸收特定波长的光的试剂和磷光体。第三系列过渡元素中,更优选的是金颗粒、铪的氧化物和钽的氧化物。镧系元素中,更优选的是镧的氧化物、铈的氧化物、镨的氧化物、钕的氧化物、铽的氧化物、镝的氧化物和镱的氧化物。通常将这些氧化物用作用于吸收特定波长的光的试剂和磷光体。
无机颗粒的颗粒尺寸可以通过各种方法测量。在本文中将颗粒尺寸的范围描述为通过使用激光的动态光散射法测量的体积基准50%累积分布直径(D50),同时可以通过电子显微镜法观察颗粒直径作为支持证据。尽管通过这样的测量方法测定的值并不取决于特定的测量仪器,可以将如Nanotrac UPA-EX150(Nikkiso Co.,Ltd.)的仪器用于动态光散射法。对于电子显微镜法,可以使用透射电子显微镜H-9500(Hitachi High-Technologies Corp.)。当将无机颗粒添加至透明有机硅材料时,例如无机颗粒的D50尺寸应当优选在1至200nm,更优选1至100nm,甚至更优选1至80nm,且最优选1至50nm的范围,因为可见光区域中的透明度至关重要。具有超过200nm的D50尺寸的颗粒大于可见光区域的波长,可能导致显著的散射。具有小于1nm的D50尺寸的颗粒可能在分散介质中产生极大的总表面积,使得它们容易聚集,并且其分散体可能难于处理。
无机颗粒的核可以为选自前述金属氧化物的一种或者两种或更多种的复合体。在此,术语“复合体”以广义使用并且是指通过简单混合或化学结合形成的复合(双或多)氧化物。通过化学结合形成的复合氧化物是指由下式(5)表示的形式。
(M1Ox)d(M2Oy)e (5)
其中,M1为选自以下的元素:Al、B、In、Si、Ge、Sn、Ti、Mn、Zn、Y、Zr、Hf、Ta、La、Ce、Pr、Nd、Tb、Dy和Yb。M2为选自以下的元素:Al、B、In、Si、Ge、Sn、Ti、Mn、Zn、Y、Zr、Hf、Ta、La、Ce、Pr、Nd、Tb、Dy和Yb,条件是M2的元素与M1的元素不相同。字母x和y给定为x=a/2,其中a为M1的价数和y=b/2,其中b为M2的价数。字母d和e为满足d+e=1,0<d<1和0<e<1的实数。即,所述结构具有其中M1经由氧与M2结合的单元。在所述结构中,M1和M2可以为稀疏分布或局部集中。在两种或更多种金属烷氧化物的共水解产物中观察到其中M1和M2为稀疏分布的结构。在核/壳型颗粒(即各自由微粒状金属氧化物的核和包封所述核的另一种金属氧化物的壳组成的颗粒)中观察到其中M1和M2为局部集中的结构,并且所述结构例如通过对应于金属烷氧化物的类型分阶段水解多种金属烷氧化物形成。
无机颗粒优选包含选自以下的至少一种化合物:氧化铝、氧化铈、氧化钛、氧化锌、氧化铟锡、氧化锆、氧化锡、氧化铁和氧化硅。在该情况下,无机颗粒优选具有核/壳结构,其具有包含选自氧化铝、氧化铈、氧化钛、氧化锌、氧化铟锡、氧化锆、氧化锡和氧化铁的至少一种化合物的核和围绕所述核的氧化硅的壳。
无机颗粒可以具有核/壳结构,其具有选自前述金属元素或金属氧化物的一种或者两种或更多种的复合体的核和围绕所述核的选自前述金属氧化物的一种或者两种或更多种的复合体的壳。典型的核/壳型颗粒为具有氧化钛-氧化锡的复合氧化物(固溶体中引入锡的颗粒状氧化钛)的核和围绕所述核的氧化硅壳的核/壳型颗粒。这样的核/壳型颗粒例如通过描述于JP 5704133中的方法制备。
接枝的聚硅氧烷的链长
优选根据归属于全部D单元的29Si-NMR信号的积分值(ΣD)与归属于全部M单元的29Si-NMR信号的积分值(ΣM)的比例、即ΣD/ΣM,来比较接枝的聚硅氧烷的链长。D单元为聚硅氧烷的骨架组成部分和M单元为聚硅氧烷的末端组成部分。据推测,ΣD/ΣM的小的值表明接枝的聚硅氧烷的链长短,而ΣD/ΣM的大的值表明接枝的聚硅氧烷的链长长。聚硅氧烷优选具有至少3、更优选至少5的ΣD/ΣM值。小于3的ΣD/ΣM值是不期望的,因为接枝的聚硅氧烷的排除体积可能过小而不能避免颗粒之间的直接接触,导致聚集。ΣD/ΣM的上限通常为至多1,000,优选至多500,尽管其并非关键。
在通过29Si-NMR光谱测量样品之前,优选将挥发物如未反应的硅氧烷和低分子量硅氧烷从样品中蒸馏除去。对于许多固体化合物而言,已知以下现象:当将固体化合物用线形聚硅氧烷或具有弱分子间相互作用的烷基链进行表面处理时,所述化合物由于化合物分子之间的相互作用降低而液化。类似地,本文中所使用的无机颗粒可以由于聚硅氧烷的接枝而液化。环状硅氧烷的开环聚合通常以平衡反应进行。当以液体形式获得无机颗粒-聚硅氧烷复合物时,液体样品可能包含一些未接枝的聚硅氧烷组分。然而,只要使用平衡反应,就很少存在或不存在ΣD/ΣM值偏离的机会,而与是否存在未接枝至颗粒表面的聚硅氧烷组分无关。因此,可以将ΣD/ΣM值作为无机颗粒-聚硅氧烷复合物的固有值用于评价。
如本文中所使用的“积分值”是指与相对于百万分之一(ppm)绘制信号强度有关的求积问题。对于求积而言,优选以特定标准通过S/N比例设定阈值。所述S/N比例为至少5,优选至少10,且更优选至少20。小于5的S/N比例可能是不适宜的,因为基线变粗并且积分的精确度恶化。可以通过Simpson法使用计算机或通过将具有代表光谱的均匀平面密度的打印介质切割成光谱轮廓并测量其重量而测定积分值。
可以将29Si-NMR光谱应用至固体或液体。在固体NMR光谱的情况下,因为必须将测量样品蒸发至干作为预处理,所以结果并不必然反映样品中的硅的结合状态。因此,优选在液体状态通过NMR光谱监测。在此时,因为29Si核具有负的旋磁比(γB),所以奥弗豪塞尔核效应(NOE)变成反转,抑制主要在共振核周围的核磁弛豫。因此,优选选择测量条件,使得负NOE可以不变得明显。在脉冲傅里叶变换NMR的情况下,该问题可以使用充足的脉冲序列来解决。例如,优选使用非共振脉冲序列。在液体29Si-NMR光谱法中,优选使用均由不含硅的材料制成的试管和探针进行分析。可以用于NMR光谱法中的示例性不含硅的材料为聚四氟乙烯,典型地为在液体29Si-NMR光谱法中,可以使用适当的弛豫剂以用于减少测量时间。作为弛豫剂,可以使用公知的试剂(例如参见Organometallics,Volume 27,Issue 4,pp500-502(2008)以及其中的引用文献)。尤其是,优选的是乙酰丙酮化铬(III),因为其完全可溶于有机溶剂并且不导致氧化钛的聚集。例如,当将几滴乙酰丙酮化铬(III)在氘代氯仿(氯仿-d3)中的浓度为约1mol/dm3的溶液用作弛豫剂时,期望地可获得弛豫效应和氘锁定效应(deuterium lock effect)。
共振磁场的标记法可以通过以百万分之一(ppm)表示以四甲基硅烷的29Si核的共振为基准的共振磁场差异而给出。根据该标记法规则,可最常在-10至-30ppm,优选-15至-25ppm的范围检测到D单元;在-5至15ppm,优选0至10ppm的范围可检测到M单元,在-5至-20ppm,优选-10至-15ppm的范围可检测到具有一个取代的烷氧基的M单元。标记法中的负值表明共振磁场在高于基准线的磁场侧具有差异。基准线的宽度取决于用于测量的NMR仪器的磁场的强度。共振线的前述优选范围为从其中应用11.75特斯拉(T)的磁场的实例获得的值。可用于NMR仪器中的磁场在5T至20T,优选8T至15T,且更优选10T至13T的范围。如果磁场小于5T,则可能因为S/N比例变得更低而难于测量。如果磁场超过20T,则可能因为共振仪器变得大尺寸而难于测量。自然而然地,本领域技术人员将会从上文给出的信息类推磁场的强度、共振线的宽度和信号的强度。
无机颗粒-聚硅氧烷复合物优选具有经由硅氧烷键连接至核结构部分的接枝结构部分。经由共价键将接枝结构部分连接至无机颗粒核结构部分对于为复合物赋予高的环境稳定性和分散稳定性有效。可以通过29Si-NMR光谱法确认核结构部分与接枝结构部分之间的硅氧烷键。
优选的是,在29Si-NMR光谱法测量中,以归属于D单元的NMR信号的改变确认核结构部分与接枝结构部分之间的硅氧烷键的存在。例如可以将归属于D'单元(其中D单元结合至聚硅氧烷的T单元)和D"单元(其中D单元结合至无机颗粒表面上的羟基)的NMR信号用于确认。通常在约-10至约-25ppm,优选约-15至约-20ppm的范围可检测到归属于D'单元(其中D单元结合至聚硅氧烷的T单元)的NMR信号。归属于D"单元(其中D单元结合至无机颗粒表面上的羟基)的NMR信号的范围随着所结合的无机颗粒的类型而变化。在氧化硅的情况下,例如通常在约-10至约-25ppm,优选约-15至约-20ppm范围可检测到归属于结合至氧化硅表面的D"单元的NMR信号。
聚硅氧烷接枝的无机氧化物优选呈液体形式或固体形式,更优选呈液体形式。固体形式的实例包括凝胶和粉末。液体形式的实例包括牛顿流体和宾汉流体,优选非触变性牛顿流体。
[包含无机颗粒-聚硅氧烷复合物的分散体]
包含无机颗粒-聚硅氧烷复合物的分散体取决于分散质的类型显示出各种功能并且提供具有功能性的油和涂料组合物。例如,当添加至涂料组合物时,分散体具有各种功能性,如,对于涂料组合物,能够赋予抗划伤性和柔性的机械功能性,能够赋予光折射控制、UV屏蔽、辐射屏蔽和荧光的光学功能性,以及能够赋予导电性和介电性能的电学功能性。包含无机颗粒-聚硅氧烷复合物的分散体可以由各种类型的元素组成并因此发挥各种功能性。然而从化工观点来看,分散体状态是类似的。当想要分散体的溶剂置换时,前述无机颗粒如金属氧化物的一种或两种或更多种的复合体可以以一组进行处理。从化工观点的状态是指在粉末工程或运输现象范畴中所讨论的物理性质,例如分散质的颗粒尺寸和ζ电势。甚至在无机颗粒-聚硅氧烷复合物的分散体包含由不同类型的元素组成的分散质时,也可以在讨论中使用这些物理量将它们确认为可比的并列组来。尽管在说明书中仅描述了包含无机颗粒-聚硅氧烷复合物的特定类型和/或组合的分散体,但也相信未描述的分散体是等价的并且落入本发明的范围。
除非另有指明,本文所使用的术语“分散体”是指稳定的分散状态(即胶体分散),颗粒并不随时间推移从其中沉降出来。有时,当颗粒的布朗运动速率快于重力沉降速度时,可以实现稳定分散状态。因为在粘性液体如有机硅中的颗粒具有慢的沉降速度,所以花费长时间来评判颗粒是否已聚集和沉淀。在这样的情况下,优选使用离心机以施加离心力来促进颗粒沉淀,因为这更有效地评判存在或不存在沉淀的颗粒。
分散质
分散体中的分散质为本文中所定义的无机颗粒-聚硅氧烷复合物。无机颗粒选自氧化硅、氧化铝、氧化钛、氧化锌、氧化铈、氧化锡、氧化硼和氧化铟,如上文所描述。通常,当将分散体添加至涂料组合物时,例如,典型地,取决于特定目的使用一种或多种类型的无机颗粒。因此不排除分散体包含多种类型的无机颗粒。将分散体添加至涂料组合物以赋予机械性质、UV屏蔽性质和导电性。为了赋予机械性质,通常使用氧化硅、氧化铝、氧化锡、氧化硼和包含至少一种这样的金属元素的复合氧化物。为了赋予UV屏蔽性质,经常使用氧化钛、氧化锌和氧化铈并且可以任选地与氧化硅、氧化铝和氧化锡(用于赋予机械性质)组合为复合体或混合物。为了赋予导电性,经常使用氧化铟-氧化锡复合体。尽管这些无机颗粒可以赋予各种功能,但是分散体的化工状态是类似的。当想要无机颗粒分散体的溶剂置换时,前述无机颗粒的一种或两种或更多种的复合体可以以一组进行处理。
分散介质
可以将各种液体用作分散体中的分散介质。实例包括包含选自以下的一种或两种或更多种的混合物的液体:C5-C30烃化合物,如戊烷、己烷、庚烷、壬烷、癸烷、十一烷、十二烷、十三烷、十四烷、十五烷、十六烷、十七烷、十八烷、十九烷、二十烷、二十一烷、二十二烷、二十三烷、二十四烷、二十五烷、二十六烷、二十七烷、二十八烷、二十九烷、三十烷、苯、甲苯、邻二甲苯、间二甲苯、对二甲苯、石油醚(或前述的混合物)、煤油、里格罗因和白润滑油;一元醇和多元醇,如甲醇、乙醇、1-丙醇、2-丙醇、环戊醇、乙二醇、丙二醇、β-硫代二甘醇、丁二醇和甘油;醚,如乙醚、丙醚、环戊基甲醚、乙二醇二甲醚、二乙二醇二甲醚、三乙二醇二甲醚、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丙醚、乙二醇单丁醚、丙二醇单甲醚、丙二醇单乙醚、丙二醇单丙醚、丙二醇单丁醚、丁二醇单甲醚、丁二醇单乙醚、丁二醇单丙醚和丁二醇单丁醚;酯,如甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丙酸丁酯、丁酸甲酯、丁酸乙酯、丁酸丙酯、丁酸丁酯、苯甲酸甲酯、苯甲酸乙酯、苯甲酸丙酯、苯甲酸丁酯、草酸二甲酯、草酸二乙酯、草酸二丙酯、草酸二丁酯、丙二酸二甲酯、丙二酸二乙酯、丙二酸二丙酯、丙二酸二丁酯、乙二醇二甲酸酯、乙二醇二乙酸酯、乙二醇二丙酸酯、乙二醇二丁酸酯、丙二醇二乙酸酯、丙二醇二丙酸酯、丙二醇二丁酸酯、乙二醇甲醚乙酸酯、丙二醇甲醚乙酸酯、丁二醇单甲醚乙酸酯、乙二醇乙醚乙酸酯、丙二醇乙醚乙酸酯和丁二醇单乙醚乙酸酯;酮,如丙酮、二丙酮醇、二乙基酮、甲基乙基酮、甲基异丁基酮、甲基正丁基酮、二丁基酮、环戊酮、环己酮、环庚酮和环辛酮;酰胺,如二甲基甲酰胺、二甲基乙酰胺、四乙酰基亚乙基二酰胺、四乙酰基六亚甲基四酰胺和N,N-二甲基六亚甲基二胺二乙酸酯;和硅氧烷,如六甲基二硅氧烷、八甲基三硅氧烷、十甲基四硅氧烷、六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环六硅氧烷、六苯基环三硅氧烷、八苯基环四硅氧烷、2,4,6,8-四甲基环四硅氧烷、1,3,5-三(3,3,3-三氟丙基)-1,3,5-三甲基环三硅氧烷、2,4,6,8-四甲基-2,4,6,8-四乙烯基环四硅氧烷、三甲基甲硅烷氧基封端的甲基氢聚硅氧烷(商品名KF-99、KF-9901,由Shin-Etsu Chemical Co.,Ltd.制)、三甲基甲硅烷氧基封端的二甲基硅氧烷(商品名KF-96-100CS、KF-96-10000CS,由Shin-Etsu Chemical Co.,Ltd.制)、三甲基甲硅烷氧基封端的甲基苯基硅氧烷、三甲基甲硅烷氧基封端的二苯基硅氧烷、二甲基乙烯基甲硅烷氧基封端的二甲基硅氧烷和二甲基氢甲硅烷氧基封端的二甲基硅氧烷,其可以单独或混合使用。要注意的是,除非另有指明,术语“封端的”意指在分子链的两端用所提及的基团将硅氧烷封端。
分散介质优选为选自以下的至少一种化合物:戊烷、己烷、庚烷、壬烷、癸烷、苯、甲苯、邻二甲苯、间二甲苯、对二甲苯和聚硅氧烷化合物。优选的聚硅氧烷化合物为选自以下的至少一种:六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷和十二甲基环六硅氧烷。聚硅氧烷优选为在25℃具有50至100,000mm2/s的动态粘度的聚二甲基硅氧烷。除非另有指明,分散介质的粘度在25℃根据JIS Z8803测量,并且聚二甲基硅氧烷的动态粘度通过奥斯特瓦尔德(Ostwald)粘度计测量。
分散体
分散体以0.001%至50重量%,更优选0.01%至30重量%的量包含无机颗粒-聚硅氧烷复合物即分散质。复合物少于0.001重量%可能不是有效浓度。包含大于50重量%的复合物的分散体可能具有差的储存稳定性。
无机颗粒-聚硅氧烷复合物与涂料组合物,典型地与有机硅涂料组合物高度相容。当将复合物引入有机涂料组合物中时,取决于无机颗粒的类型其对涂料组合物赋予各种功能性。所述功能性的实例包括能够赋予抗划伤性和柔性的机械功能,能够赋予光折射控制、UV屏蔽、辐射屏蔽和荧光的光学功能,和能够赋予导电性和介电性能的电学功能。
[固体材料]
除非另有指明,术语“固体材料”是指在标准环境温度和压力下是稳定的并具有某种形状和体积的材料,并且不根据形式如玻璃、橡胶、结晶或无定形形式对其区分。
可以将无机颗粒-聚硅氧烷复合物与各种有机树脂基材混合,以形成其中均匀地分散有无机颗粒的的树脂材料。有机树脂的实例包括聚碳酸酯、聚苯乙烯、丙烯酸系树脂、改性的丙烯酸系树脂、聚氨酯树脂、硫代聚氨酯树脂、卤化的双酚A与乙二醇的缩聚物、丙烯酸系聚氨酯树脂、卤化的含芳基的丙烯酸系树脂、含硫的树脂、硅橡胶和有机硅树脂。此外,无机颗粒-聚硅氧烷复合物能够取决于无机颗粒的类型而将各种功能性赋予树脂。所述功能的实例包括能够改进硬度、拉伸强度和耐热性的机械功能性,能够赋予光折射控制、UV屏蔽、辐射屏蔽和荧光的光学功能性,和能够赋予导电性和介电性能的电学功能性。
可以通过任意期望的方法将无机颗粒-聚硅氧烷复合物与各种有机树脂基础混合,所述方法例如为在树脂的聚合或固化步骤之前混合复合物的方法,或直接捏合复合物与树脂的方法。为了将无机颗粒充分地分散在树脂中优选在树脂的聚合或固化步骤之前混合复合物的方法。
[硅橡胶材料]
除非另有指明,术语“硅橡胶材料”是指显示出橡胶弹性的材料,在标准环境温度和压力下是稳定的并具有某种形状和体积,优选具有在低于25℃范围内的玻璃化转变温度的材料。
硅橡胶材料以优选0.001至20重量%、更优选0.01至20重量%的浓度包含无机颗粒-聚硅氧烷复合物。如果复合物的含量少于0.001重量%,则复合物(无机颗粒)可能无法发挥其功能性。多于20重量%的复合物可能减损硅橡胶的性质。
包含无机颗粒-聚硅氧烷复合物的硅橡胶材料的特征在于高的可见光透射性。可见光透射性的一个指标为橡胶材料在可见光区域中的光透射率。因为膜的可见光透射率随着膜厚度增加而降低,所以将具有1mm厚度的橡胶材料样品用作标准。优选在500nm波长具有的光透射率相较于不含复合物的有机硅材料的光透射率不经历至少10%、更优选不经历至少5%的降低的负载有复合物的有机硅材料样品。值得注意的是,橡胶材料的光透射率可以通过UV/Vis吸收光谱仪测量。
可见光透射率的另一指标为雾度(Hz)。因为膜的雾度随着膜厚度增加而增加,所以将具有1mm厚度的橡胶材料样品用作标准。优选雾度相较于不含复合物的有机硅材料的雾度不经历至少20%、更优选不经历至少10%的增加的负载有复合物的有机硅材料样品。值得注意的是,橡胶材料的雾度可以通过浊度计测量。
本文中所使用的硅橡胶材料可以基于任意公知的有机聚硅氧烷。典型地,所述有机聚硅氧烷具有平均组成式(6):
R51fSiO(4-f)/2 (6)
其中,R51各自独立地为取代或未取代的一价烃基和f为1.98至2.02的正数。
在式(6)中,R51各自独立地为取代或未取代的一价烃基,优选具有1至10个碳原子,更优选1至8个碳原子。合适的取代或未取代的一价烃基包括烷基,如甲基、乙基、异丙基、丁基、异丁基、叔丁基、己基和辛基;环烷基,如环己基;烯基,如乙烯基和烯丙基;芳基,如苯基和甲苯基;芳烷基,如苄基、苯乙基和苯基丙基;和前述基团的一些或全部氢原子被卤原子、氰基等取代的取代形式,其中,例如卤素取代的和氰基取代的烷基,如氯甲基、溴乙基和氰基乙基。尤其优选甲基、乙烯基、苯基和三氟丙基。更优选地,甲基占R51基团的至少50mol%,特别是至少80mol%。下标f为1.98至2.02的正数。优选的是每分子包含至少两个烯基的那些有机聚硅氧烷,特别是0.001至5mol%的R51基团为烯基。
尽管具有式(9)的有机聚硅氧烷的分子结构没有特别限制,但优选的是在分子链末端用三有机基甲硅烷基,特别是二有机基乙烯基甲硅烷基(例如二甲基乙烯基甲硅烷基)封端的有机聚硅氧烷。基本上,有机聚硅氧烷优选具有线形结构,尽管不同分子结构的混合物是可接受的。
有机聚硅氧烷优选具有100至100,000、更优选100至2,000的平均聚合度(DOP)。另外,有机聚硅氧烷优选在25℃具有100至100,000,000mm2/s,更优选100至100,000mm2/s的粘度。要注意的是,平均DOP是基于通过GPC相对于聚苯乙烯标样的重均分子量的值;粘度在25℃根据JIS Z8803测量。
在一个实施方案中,所述硅橡胶材料为有机过氧化物固化类型。在该实施方案中,将有机过氧化物用作固化剂。当作为基础的有机聚硅氧烷为具有至少3,000的DOP的胶状的有机聚硅氧烷时,有机过氧化物固化是有效的。可以使用任意公知的有机过氧化物,例如过氧化苯甲酰、过氧化2,4-二氯代苯甲酰、过氧化对甲基苯甲酰、过氧化邻甲基苯甲酰、过氧化2,4-二异丙苯基、2,5-二甲基-2,5-双(叔丁基过氧基)己烷、二叔丁基过氧化物、过氧化苯甲酸叔丁酯、1,1-双(叔丁基过氧基)-3,3,5-三甲基环己烷、和1,6-双(叔丁基过氧羧基)己烷。有机过氧化物优选以0.01至10重量份的量添加,按每100重量份的有机聚硅氧烷计。此外,还可以将任意公知的添加剂配混至硅橡胶材料中。
在另一实施方案中,硅橡胶材料为加成反应固化类型。该实施方案使用每分子包含至少两个烯基(典型地,乙烯基)的有机聚硅氧烷作为前述有机聚硅氧烷,有机氢聚硅氧烷作为交联剂和加成反应催化剂作为固化剂。
有机氢聚硅氧烷优选在常温为液体并具有平均组成式(7)。
R52gHhSiO(4-g-h)/2 (7)
其中,R52各自独立地为C1-C10取代或未取代的一价烃基,g为满足以下范围的数:0≤g≤3,优选0.7≤g≤2.1;h为满足以下范围的数:0<h≤3,优选0.001≤h≤1;和0<g+h≤3,优选0.8≤g+h≤3.0。
在式(7)中,R52为C1-C10,优选C1-C8取代或未取代的一价烃基,如上文对R51所示例,典型地不含脂族不饱和度。示例性基团包括烷基、芳基、芳烷基和被取代的烷基,如甲基、乙基、丙基、苯基和3,3,3-三氟丙基。
有机氢聚硅氧烷的分子结构可以为线形、环状、支链或三维网络,同时SiH基团可以位于分子链的末端或中途位置或这二者处。尽管分子量没有特别限制,有机氢聚硅氧烷优选在25℃具有1至1,000mm2/s,特别是3至500mm2/s的粘度。
有机氢聚硅氧烷的实例包括
1,1,3,3-四甲基二硅氧烷、甲基氢环聚硅氧烷、
甲基氢硅氧烷/二甲基硅氧烷环状共聚物、
三甲基甲硅烷氧基封端的甲基氢聚硅氧烷、
三甲基甲硅烷氧基封端的二甲基硅氧烷/甲基氢硅氧烷共聚物、
二甲基氢甲硅烷氧基封端的二甲基聚硅氧烷、
二甲基氢甲硅烷氧基封端的二甲基硅氧烷/甲基氢硅氧烷共聚物、
三甲基甲硅烷氧基封端的甲基氢硅氧烷/二苯基硅氧烷共聚物、
三甲基甲硅烷氧基封端的甲基氢硅氧烷/二苯基硅氧烷/二甲基硅氧烷共聚物、
由(CH3)2HSiO1/2单元和SiO4/2单元组成的共聚物、
由(CH3)2HSiO1/2单元、(CH3)3SiO1/2单元和SiO4/2单元组成的共聚物,和
由(CH3)2HSiO1/2单元、SiO4/2单元和(C6H5)3SiO1/2单元组成的共聚物。
要注意的是,术语“封端的”意指在分子链的两端用所提及的基团将硅氧烷封端。
有机氢聚硅氧烷优选以这样的量使用,使得在基础聚合物中每个硅结合的烯基可获得0.1至10个,更优选0.2至2个硅结合的氢原子(SiH基团)。
加成反应催化剂选自铂族金属催化剂,其包括单独的铂族金属及其化合物和络合物。示例性铂族金属催化剂包括铂催化剂如铂黑、氯化铂、氯铂酸、氯铂酸与一元醇的反应产物、氯铂酸与烯烃的络合物、和双乙酰乙酸铂,钯催化剂如四(三苯基膦)合钯和二氯化双(三苯基膦)合钯,和铑催化剂如氯化三(三苯基膦)合铑和四(三苯基膦)合铑。
加成反应催化剂可以以催化量使用,其典型地为0.1ppm至1,000ppm,更优选1至200ppm的铂族金属,基于含烯基的有机聚硅氧烷的重量计。少于0.1ppm可能不足以促进加成反应或固化,而大于1,000ppm可能是不经济的。
前述类型中,加成反应类型的硅橡胶组合物是优选的,因为其通常固化成低硬度产物。
在硅橡胶材料中,如果需要并且只要不影响本发明的目的,则可以添加各种其它组分。合适的另外的组分示于下文,同时每种组分可以为一种或多种的混合物。
例如,可以将加成反应抑制剂共混于硅橡胶材料中以提供适用期。加成反应抑制剂没有特别限制,只要其将固化抑制效果赋予氢化硅烷化催化剂。加成反应抑制剂可以选自常规公知化合物,例如含磷化合物如三苯基膦,含氮化合物如三丁胺、四甲基乙二胺和苯并三唑,含硫化合物,炔属化合物如炔属醇(例如1-乙炔基环己醇、3,5-二甲基-1-己炔-3-醇),包含至少两个烯基的化合物,氢过氧化合物,和马来酸衍生物。
加成反应抑制剂的固化抑制效果的程度随着抑制剂的化学结构而变化。因此,每当选择特定的加成反应抑制剂时,优选单独地调节其量为最优化。当添加最优化的量的加成反应抑制剂时,硅橡胶组合物在室温长期货架储存期间保持稳定并且仍然有效地热固化。
可以将任意公知的抗氧化剂如2,6-二-叔丁基-4-甲基苯基添加至硅橡胶组合物以防止组合物着色、混浊和氧化降解。可以将光稳定剂如受阻胺稳定剂添加至硅橡胶组合物以赋予对光降解的耐受性。可以将无机填料如气相二氧化硅添加至硅橡胶组合物以改进强度,只要不负面地影响组合物的固化的产物的透明度。如果需要,可以将染料、颜料和阻燃剂添加至硅橡胶组合物。
[用于制备无机颗粒-聚硅氧烷复合物的方法]
尽管如本文中所定义的用于制备无机颗粒-聚硅氧烷复合物的方法并没有特别限制,但是以下描述的方法是优选的。本发明的另一实施方案是用于制备无机颗粒-聚硅氧烷复合物的方法,其包括以下步骤:
(a)提供无机颗粒在极性有机溶剂中的分散体,所述无机颗粒具有通过动态光散射法测量的1至200nm的体积基准50%累积颗粒尺寸,和
(b)混合所述分散体与至少一种具有通式(1)的环状硅氧烷,并使混合物中的所述环状硅氧烷进行开环聚合。
其中,R7、R8、R9、R10、R11和R12各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代,和n为0至10的整数。
步骤(a)
步骤(a)为提供无机颗粒在极性有机溶剂中的分散体,所述无机颗粒具有通过动态光散射法测量的1至200nm的体积基准50%累积颗粒尺寸。
除非另有指明,如上文所提及,术语“分散体”是指稳定的分散状态(即胶体分散体),颗粒并不随时间推移从其中沉降出来。当颗粒的布朗运动速率快于重力沉降速度时,可以实现稳定分散状态。因为在粘性液体如有机硅中的颗粒具有慢的沉降速度,所以难于评判分散状态是否稳定。在这样的情况下,可以使用离心机以施加离心力来促进颗粒沉淀,因为这会加快评判存在或不存在沉淀的颗粒。
在步骤(a)中提供的无机颗粒如上文定义。起始无机颗粒可以为合成产品或商购可得的产品。商购可得的产品的实例包括IPA-ST-L(二氧化硅纳米颗粒的IPA分散体,Nissan Chemical Industries,Ltd.),氧化铝纳米颗粒的IPA分散体(Sigma-Aldrich),氧化锌的乙酸丁酯分散体(Sigma-Aldrich),氧化铟锡的IPA分散体(Sigma-Aldrich)和ITRANB15WT%-G180(氧化铟锡的醇分散体,CIK NanoTech)。还包括Tainok M-6(二氧化钛水分散体,Taki Chemical Co.,Ltd.),Needral P10(氧化铈水分散体,Taki Chemical Co.,Ltd.),Nanouse CE-T20B(氧化铈水分散体,Nissan Chemical Industries,Ltd.),Snowtex ST-20L(二氧化硅水分散体,Nissan Chemical Industries,Ltd.),AS-100(氧化铝水分散体,Nissan Chemical Industries,Ltd.),Bayral AlML15(氧化铝水分散体,Taki Chemical Co.,Ltd.),ALW10WT%-G0(氧化铝水分散体,CIK NanoTech),CEW10WT%-G120(氧化铈水分散体,CIK NanoTech),Cemeral S8(氧化锡水分散体,Taki Chemical Co.,Ltd.),Bayral Fe-C10(氧化铁水分散体,Taki Chemical Co.,Ltd.),氧化锆水分散体(Sigma-Aldrich),ITRW15WT%-G30(氧化铟锡水分散体,CIK NanoTech),条件是在使用前作为分散介质的水可以被极性有机溶剂置换。另外,在溶剂置换之前可以将这些水分散体用常规硅烷偶联剂或分散剂进行表面处理。
起始胶体分散体
步骤(a)优选使用无机氧化物颗粒在作为分散介质的水中的胶体分散体。水可以为淡水,如自来水、工业用水、井水、自然水、雨水、蒸馏水或去离子水,优选的是去离子水。去离子水可以通过纯水生产单元例如由Organo Corp.制造的FW-10或由Merck Millipore Corp.制造的Direct-QUV3生产。分散介质可以包含水可混溶性一元醇,所述一元醇在制备下文待描述的经水分散的胶体溶液的步骤期间引入。还可以包含水可混溶性一元醇,其源自用于制备核/壳颗粒的共溶剂和金属烷氧化物上的溶胶-凝胶反应的水解副产物。
在步骤(a)中提供的起始胶体分散体优选具有1至35重量%,更优选5至30重量%,且甚至更优选10至25重量%的分散质浓度。小于1重量%的分散质浓度因为低生产效率而是不期望的。大于35重量%的分散质浓度是不期望的,因为在某些条件如pH和温度下可能发生胶凝。
无机颗粒在其表面上具有SiR1O3/2单元的硅氧烷涂覆层的情况下,步骤(a)包括将无机颗粒用硅烷化合物和/或其(部分)水解缩合物进行表面处理的步骤(a-1)和用极性有机溶剂置换溶剂的步骤(a-2)。无机颗粒在其表面上不具有SiR1O3/2单元的硅氧烷涂覆层的情况下,步骤(a)仅为步骤(a-2)。
步骤(a-1)
步骤(a-1)为将具有通式(I)的硅烷化合物和/或其(部分)水解缩合物添加至无机颗粒,由此将颗粒表面改性。
R19Si(Y)3 (I)
其中,R19各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基、氨基、巯基、异氰酸酯基团或氟基团取代。Y为选自烷氧基、乙酰氧基、烯醇和氯的基团。
合适的烷基为C1-C6烷基,典型地甲基和乙基。合适的芳基为C6-C10芳基,典型地为苯基。合适的(聚)二甲基甲硅烷氧基具有1至50个硅原子,特别是1至30个硅原子。
具有式(I)的硅烷化合物的实例为烷氧基硅烷,包括氢三甲氧基硅烷、氢三乙氧基硅烷、甲基三甲氧基硅烷、甲基三乙氧基硅烷、甲基三异丙氧基硅烷、乙基三甲氧基硅烷、乙基三乙氧基硅烷、乙基三异丙氧基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、丙基三异丙氧基硅烷、苯基三甲氧基硅烷、乙烯基三甲氧基硅烷、烯丙基三甲氧基硅烷、γ-甲基丙烯酰氧基丙基三甲氧基硅烷、γ-甲基丙烯酰氧基丙基三乙氧基硅烷、γ-丙烯酰氧基丙基三甲氧基硅烷、γ-缩水甘油氧基丙基三甲氧基硅烷、γ-缩水甘油氧基丙基三乙氧基硅烷、β-(3,4-环氧基环己基)乙基三甲氧基硅烷、γ-氯丙基三甲氧基硅烷、3,3,3-三氟丙基三甲氧基硅烷、3,3,3-三氟丙基三乙氧基硅烷、全氟辛基乙基三甲氧基硅烷、γ-巯基丙基三甲氧基硅烷、γ-氨基丙基三甲氧基硅烷、γ-氨基丙基三乙氧基硅烷、N-(2-氨基乙基)氨基丙基三甲氧基硅烷、γ-异氰酸酯基丙基三甲氧基硅烷、γ-异氰酸酯基丙基三乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、其中异氰酸酯基团结合在一起的三(3-三甲氧基甲硅烷基丙基)异氰脲酸酯和三(3-三乙氧基甲硅烷基丙基)异氰脲酸酯;和甲基三甲氧基硅烷的部分水解缩合物,其以KC-89S和X-40-9220商业上可获自Shin-Etsu Chemical Co.,Ltd.;和γ-缩水甘油氧基丙基三甲氧基硅烷和甲基三甲氧基硅烷的部分水解缩合物,它们以X-41-1056商业上可获自Shin-Etsu Chemical Co.,Ltd.。还包括烯丙基硅烷,如三烯丙基甲基硅烷、三烯丙基乙基硅烷和三烯丙基异丙基硅烷;乙酰氧基硅烷如三乙酰氧基甲基硅烷、三乙酰氧基乙基硅烷、三乙酰氧基丙基硅烷和三乙酰氧基苯基硅烷;氯硅烷,如三氯甲基硅烷、三氯乙基硅烷、三氯丙基硅烷和三氯苯基硅烷;和烯醇硅烷如,三异丙烯基氧基甲基硅烷、乙基三异丙烯基氧基硅烷、三异丙烯基氧基丙基硅烷和苯基三异丙烯基氧基硅烷。
其中R19为(聚)二甲基硅氧烷的具有式(I)的硅烷化合物的实例包括具有通式(II)的化合物。
在式(II)中,r优选为0至50的整数,更优选5至40的整数,且甚至更优选10至30的整数。如果r大于50,则化合物不期望地可能在硅油特性方面变得更强,使得经表面处理的有机溶胶在各种树脂中的溶解性受到限制。具有其中r=30的平均结构的式(II)的化合物商业上以X-24-9822可获自Shin-Etsu Chemical Co.,Ltd.。
所添加的硅烷化合物的量优选为起始胶体水分散体的固体重量的0.5至50倍,更优选1至25倍,且甚至更优选2至10倍。如果所添加的硅烷化合物的量大于起始胶体水分散体的固体重量的50倍,则可能发生胶凝。如果所添加的硅烷化合物的量小于起始胶体水分散体的固体重量的0.5倍,则可能由于缺少被覆而发生聚集。
可以通过任意技术如在液体中滴加、在液体外滴加和分批滴加来添加硅烷化合物,优选的是在液体中滴加。
在步骤(a-1)中,如果需要可以添加用于促进表面处理的酸催化剂或碱催化剂。合适的碱催化剂包括氢氧化钾、氢氧化钠、碳酸钾、碳酸钠和碱性离子交换树脂。合适的酸催化剂包括盐酸、硫酸、甲磺酸、三氟甲磺酸、乙酸和阳离子交换树脂。阳离子交换树脂的实例包括(Organo)、(Lanxess)、(Purolite)和(Muromachi Chemicals Inc.)。催化剂优选以0.01至20重量%,更优选0.1至10重量%,且甚至更优选1至5重量%的量使用,基于无机颗粒的重量计。如果催化剂的量大于20重量%,则不期望地发生快速反应并且难于控制。如果催化剂的量少于0.01重量%,则不期望地几乎不发生反应。
当添加硅烷化合物时,胶体水分散体优选在0至45℃,更优选5至40℃,且甚至更优选10至35℃的温度。在低于0℃的温度,分散体可能不期望地经由因冷冻引起的状态改变而变化。在高于45℃的温度,添加的硅烷可能不期望地经历意外的水解缩合,并且由于水解缩合,反应溶液可能升高至低于70℃的更高的温度。
在步骤(a-1)中,如果需要,可以将反应溶液用有机溶剂稀释。合适的稀释用溶剂包括一元醇如甲醇、乙醇、异丙醇和丁醇;多元醇如乙二醇、丙二醇和甘油;醚如丙二醇单甲醚、乙二醇单甲醚、乙二醇二甲醚和二乙二醇二甲醚;酮如丙酮和甲基异丁基酮;酯如乙酸乙酯和丙二醇单甲醚乙酸酯(PGMEA);和反应性酯如己二醇二丙烯酸酯、三羟甲基丙烷三丙烯酸酯、季戊四醇四丙烯酸酯和二季戊四醇六丙烯酸酯。尤其是,优选的是乙醇和异丙醇。优选进行稀释,以避免在随后的步骤(a-2)中的溶剂冲击(solvent shock),但并非必要。稀释率优选为1至20倍,更优选2至15倍,且甚至更优选3至10倍。小于1的稀释率可能太低而不能发挥想要的溶剂冲击减轻效果。如果稀释率大于20倍,则随后的步骤可能花费不必要的长的处理时间。
步骤(a-2)
步骤(a-2)为用极性有机溶剂置换反应溶液的分散介质。如果需要,则可以通过超滤浓缩分散体以使分散介质渗出。分散介质可以包含在步骤(a-1)中制备的水分散体中所含的水、来源于所添加的硅化合物和/或其水解缩合物和/或通过水解缩合形成的硅酸酯的醇、和有机溶剂。通过使这样的复杂体系分散体渗出,可以将过滤室中的分散体浓缩至优选1至30重量%,更优选5至25重量%,且甚至更优选10至20重量%的固体浓度。虽然本文中待渗出的分散介质为复杂的混合物,但有利地使用多孔陶瓷过滤器。尽管常规技术推荐将中空纤维膜用于从水中除去盐,但是中空纤维膜可能在细颗粒分散体通过时被堵塞。在涉及除去细颗粒、浓缩和固液分离的区域中,经常使用有机聚合物系超滤膜。然而,如果包含有机溶剂,则过滤膜将会溶胀并且变得不能使用。因此,对于固液分离和含有机溶剂的样品的浓缩而言,无机陶瓷过滤器是有效的。
对于超滤而言,使用包括具有优选5nm至小于20nm,更优选6nm至小于18nm,且最优选7nm的平均孔尺寸的无机陶瓷膜的过滤器。过滤器优选为可旋转盘。无机陶瓷膜可以通过任意公知的技术制备。制成无机陶瓷膜的材料包括尖晶石、氧化铝、二氧化钛和氧化锆基材料。例如,尖晶石系材料可以通过已知技术合成(Ueno等.,Journal of Physics:Conference Series 2009,Vol.165,No.1,Fabrication of porous magnesium spinel with cylindrical pores by unidirectional solidification或Zhang,Guo-Chang,等.,2000,Vol.2000,No.03,MgAl2O Ultrafiltration Ceramic Membrane Derived from Mg-Al Double Alkoxide)。优选地,通过调节尖晶石晶体的生长和合成条件控制孔尺寸。优选通过在多孔盘状未上釉的氧化铝等的陶瓷板上通过外延生长沉积具有均匀的孔尺寸的表面层形成过滤器。本文中所使用的氧化铝多孔盘典型地为具有0.05至1μm的孔尺寸的那种。表面层具有优选5nm至小于20nm,更优选6nm至小于18nm,且最优选7nm的平均孔尺寸。关于盘状过滤器的尺寸,其直径优选为100mm至小于500mm,更优选120mm至小于300mm,且甚至更优选140mm至200mm。如果直径小于100mm,则在旋转时不期望地几乎不施加剪切应力并且不保证一定的表面积。如果直径超过500mm,则可能需要附加扭矩来旋转。具有过大直径的过滤器还倾向于破裂并且难于处理。过滤器的厚度优选为1mm至小于10mm,更优选3mm至小于5mm。具有小于1mm厚度的过滤器可能缺乏机械强度,而具有超过10mm厚度的过滤器可能在提供具有一定体积的过滤室上不利。可以通过公知的技术制造过滤器或可以使用商购可得的过滤器。
用于溶剂置换步骤的过滤器的孔尺寸优选通过电子显微镜法测定。用于此目的的电子显微镜可以为扫描电子显微镜、透射电子显微镜或原子力显微镜。
在溶剂置换步骤中,在优选小于0.5MPa,更优选小于0.4MPa,甚至更优选小于0.3MPa,且最优选小于0.2MPa和至少0.03MPa的静压力下使分散介质渗出。如果静压力等于或大于0.5MPa,则用于超滤系统的界面的选择可能受到限制。低于0.03MPa的静压力可能在高效渗出上失效。优选使用表面与空气接触或为封闭体系的液压头管通过液压压力或压缩气动压力实现静压力。特别是,压缩气动压力系统因为单元紧凑而是优选的。可以通过任意公知的技术或商购可得的压缩机容易地生产压缩空气。
在溶剂置换步骤中,将优选0.1至10Pa,更优选0.5至5Pa,且甚至更优选1至5Pa的剪切应力经由过滤器施加至分散体。可以通过分散体的流动或通过盘状过滤器的旋转实现剪切应力。当通过过滤器的旋转实现剪切应力时,期望地在过滤器表面可获得高剪切率。可以由过滤室中的壁到壁的距离和旋转速度计算剪切应力。如果需要,过滤室可以配备有适当的挡板。出于减小过滤室中的壁到壁的距离的目的优选安装挡板。公知的实践是通过利用旋转和挡板增加剪切应力。可以例如根据方程式(1)计算作用在圆周上的最大剪切应力(τ):
τ=(η·π·φ·ω)/L [Pa] 方程式(1)
其中,φ为盘状过滤器的直径(m),ω为过滤器的旋转速率(rps),L为过滤器与过滤室之间的壁到壁的距离(m),π为圆周率和η为分散体的粘度(Pa·s)。假设一个实例,其中直径φ=0.15m,过滤器旋转速率ω=16.7rps圆周率π=3.14,分散体粘度η=0.001Pa·s和壁距离L=0.003m,则可以通过改变参数φ、ω和L控制剪切应力以落入优选的范围。
在溶剂置换步骤中施加至分散体的旋转能量优选由剪切应力规定,但也可以由流体状态规定。流体状态可以由雷诺数规定。搅拌雷诺数优选为3,000至5,000,000,更优选5,000至1,000,000,且甚至更优选10,000至500,000。小于3,000的雷诺数表明层流搅拌并且因此表明难于有效分散,而大于5,000,000的雷诺数从工业效率方面来看可能是不利的,因为搅拌要求不必要的大量能量。要注意的是,可以由方程式(2)确定雷诺数(Re):
Re=ρ·ω·φ2/η 方程式(2)
其中,ρ为密度(kg/m3),ω为旋转速率(rps),φ为过滤器直径(m)和η为粘度(Pa·s)。
本文中所使用的无机颗粒分散体通常具有900至2,000kg/m3,优选1,000至1,500kg/m3的密度ρ和0.001至0.05Pa·s,优选0.002至0.01Pa·s的粘度η。例如,当将ρ=1,000kg/m3和η=0.001Pa·s的氧化钛分散体通过具有φ=0.15m的盘状过滤器以ω=16.7rps处理时,计算Re为~3.8×105。可以通过适当选择ω和φ而将Re调节至落入期望的范围。对于搅拌,可以实行使用配备有挡板的反应器的改进搅拌效率的方法。
溶剂置换步骤优选在5至80℃,更优选10至60℃,甚至更优选15至50℃,且最优选20至40℃的温度进行。低于5℃,则分散体可能冻结。高于80℃的温度导致分散介质挥发掉,对加工环境产生问题,和/或将反应能量提供至反应性酯(如果使用的话),导致胶凝。通常,分散体的粘度取决于温度。因为粘度影响转矩,所以溶剂置换步骤可以在调节温度的同时进行,使得可以不将任意额外负荷施加至电磁旋转机和/或发动机。
在溶剂置换步骤中,如果需要,还可以通过连续超滤除去未反应的化合物和副产物。
有机溶剂的实例包括一元醇和多元醇,如甲醇、乙醇、1-丙醇、2-丙醇、环戊醇、乙二醇、丙二醇、β-硫代二甘醇、丁二醇和甘油;醚,如乙醚、丙醚、环戊基甲基醚、乙二醇二甲醚、二乙二醇二甲醚、三乙二醇二甲醚、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丙醚、乙二醇单丁醚、丙二醇单甲醚、丙二醇单乙醚、丙二醇单丙醚、丙二醇单丁醚、丁二醇单甲醚、丁二醇单乙醚、丁二醇单丙醚和丁二醇单丁醚;酯,如甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丙酸丁酯、丁酸甲酯、丁酸乙酯、丁酸丙酯、丁酸丁酯、苯甲酸甲酯、苯甲酸乙酯、苯甲酸丙酯、苯甲酸丁酯、草酸二甲酯、草酸二乙酯、草酸二丙酯、草酸二丁酯、丙二酸二甲酯、丙二酸二乙酯、丙二酸二丙酯、丙二酸二丁酯、乙二醇二甲酸酯、乙二醇二乙酸酯、乙二醇二丙酸酯、乙二醇二丁酸酯、丙二醇二乙酸酯、丙二醇二丙酸酯、丙二醇二丁酸酯、乙二醇甲醚乙酸酯、丙二醇甲醚乙酸酯、丁二醇单甲醚乙酸酯、乙二醇乙醚乙酸酯、丙二醇乙醚乙酸酯和丁二醇单乙醚乙酸酯;酮,如丙酮、二丙酮醇、二乙基酮、甲基乙基酮、甲基异丁基酮、甲基正丁基酮、二丁基酮、环戊酮、环己酮、环庚酮和环辛酮;酰胺,如二甲基甲酰胺、二甲基乙酰胺、四乙酰基亚乙基二酰胺、四乙酰基六亚甲基四酰胺和N,N-二甲基六亚甲基二胺二乙酸酯。这些之中,为了无机颗粒的分散和易于蒸馏分散介质,优选的是甲醇、乙醇、异丙醇和正丙醇。
用于溶剂置换步骤的有机溶剂的体积优选为过滤室体积的1至20倍,更优选2至10倍,且甚至更优选3至8倍。小于1倍的体积可能不足以用于溶剂置换,而大于20倍体积从工业效率方面来看可能是不期望的。
无机颗粒在其表面上不具有SiR1O3/2单元的硅氧烷涂覆层的情况下,步骤(a-1)是不必要的并且仅进行步骤(a-2)。在该情况下,当将无机颗粒作为在水中的分散体提供时,如上文所描述进行溶剂置换。当以粉末形态提供无机颗粒时,它们可以分散在有机溶剂中。当将无机颗粒作为在有机溶剂中的分散体提供时,步骤(a-2)是不必要的。
步骤(b)
步骤(b)包括使与由步骤(a)得到的无机颗粒分散体混合的环状硅氧烷进行开环聚合。
通常,对于采用接枝聚合物对颗粒的表面改性,已知两种方法,一种方法为将预先增加分子量的聚合物取代至颗粒表面和一种方法为从颗粒表面作为起始点生长聚合物。前一方法难于使聚合物以高密度接枝至颗粒表面,因为聚合物随着分子量增加其在端部的反应性降低。在如本发明中所使用的具有相对高反应性的低分子量硅氧烷的共存下,从颗粒表面作为起始点生长聚合物的后一方法易于以高密度将颗粒表面改性,因为一旦单体接枝至颗粒表面,其就生长成高分子量形式。此外,因为聚合反应在颗粒表面上发生,在颗粒改性之前合成聚合物是不必要的。那么,后一方法在操作效率方面优于前一方法。
步骤(a)中提供的无机颗粒分散体具有优选1至30重量%,更优选2至20重量%,且甚至更优选5至10重量%的分散质浓度。小于1重量%的分散质浓度可能导致低的加工效率,而大于30重量%的分散质浓度可能导致颗粒在复合步骤中聚集在一起。
环状硅氧烷
在用于制备无机颗粒-聚硅氧烷复合物的方法中,进行开环聚合的环状硅氧烷为至少一种具有通式(1)的环状硅氧烷。
其中,R7、R8、R9、R10、R11和R12各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代,n为0至10的整数。
在式(1)中,R7至R12如上文对R2至R6所示例。环状硅氧烷的实例包括六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环六硅氧烷、六苯基环三硅氧烷、八苯基环四硅氧烷、2,4,6,8-四甲基环四硅氧烷、1,3,5-三(3,3,3-三氟丙基)-1,3,5-三甲基环三硅氧烷、1,3,5-三甲基-1,3,5-三乙烯基环三硅氧烷和2,4,6,8-四甲基-2,4,6,8-四乙烯基环四硅氧烷。尤其优选的是选自以下的至少一种硅氧烷:六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、十二甲基环六硅氧烷和2,4,6,8-四甲基环四硅氧烷。
以优选10至1,000重量份,更优选50至1,000重量份,且甚至更优选100至500重量份的量添加环状硅氧烷,按照每100重量份的无机颗粒计。如果按照每100重量份的无机颗粒计,环状硅氧烷的量少于10重量份,则表面处理可能不期望地不足,可能聚集。
环状硅氧烷的开环聚合在优选25至150℃,更优选40至130℃,且甚至更优选60至100℃的液体温度进行。在低于25℃的液体温度,开环聚合和接枝至无机颗粒可能不充分地进行。在高于150℃的液体温度可能导致环状硅氧烷蒸发掉并从反应体系中消失。
可以将酸催化剂或碱催化剂用于促进无机颗粒与聚硅氧烷之间的复合反应的目的。合适的酸催化剂包括乙酸、盐酸、硫酸、甲磺酸、三氟甲磺酸和对甲苯磺酸。合适的碱催化剂包括氢氧化钠、氢氧化钾、甲醇钠、乙醇钠、甲醇钾、乙醇钾和硅醇钾(potassium siliconate)。适当的催化剂必须取决于环状硅氧烷的类型而选自这些化合物。例如,当具有氢甲硅烷基的环状硅氧烷为反应物时,酸催化剂是足够的。所添加的催化剂的量优选为10至0.01重量%,更优选5至0.1重量%,基于环状硅氧烷计,但不限于此。
如果添加的话,可以通过合适的手段,如用碱或酸中和或通过具有离子捕获能力的无机氧化物吸附从体系中除去酸催化剂或碱催化剂。
在步骤(b)中,环状硅氧烷的开环聚合可以在作为聚合物扩链(D单元)组分的具有通式(2)的硅烷化合物和/或其水解缩合物存在下进行:
R13R14Si(OR15)2 (2)
其中,R13和R14各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代,R15为氢或具有至多6个碳原子的烷基。其后,蒸馏除去分散介质,生成无机颗粒-聚硅氧烷复合物。所添加的具有式(2)的硅烷化合物的量优选为至多1摩尔当量,更优选至多0.5摩尔当量,相对于环状硅氧烷计。大于1摩尔当量的硅烷化合物因为聚合的进行和因此可能妨碍接枝而是不期望的。聚合物扩链组分的共存使得能够将各种官能团引入至聚硅氧烷链上,这使得折射控制、与分散介质的相容性上的改进、粘度控制或引入反应性侧链成为可能。要注意的是,当使用具有式(2)的硅烷化合物和/或其水解缩合物时,所添加的硅烷化合物的量相对于环状硅氧烷优选为至少0.01摩尔当量,更优选至少0.05摩尔当量。
具有式(2)的硅氧烷化合物的实例包括二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、甲基乙基二甲氧基硅烷、二乙基二甲氧基硅烷、二乙基二乙氧基硅烷、甲基丙基二甲氧基硅烷、甲基丙基二乙氧基硅烷、二异丙基二甲氧基硅烷、苯基甲基二甲氧基硅烷、苯基甲基二乙氧基硅烷、二苯基二甲氧基硅烷、二苯基二甲氧基硅烷、乙烯基甲基二甲氧基硅烷、γ-缩水甘油氧基丙基甲基二甲氧基硅烷、γ-缩水甘油氧基丙基甲基二乙氧基硅烷、β-(3,4-环氧基环己基)乙基甲基二甲氧基硅烷、γ-甲基丙烯酰氧基丙基甲基二甲氧基硅烷、γ-甲基丙烯酰氧基丙基甲基二乙氧基硅烷、γ-巯基丙基甲基二甲氧基硅烷、γ-氨基丙基甲基二乙氧基硅烷、N-(2-氨基乙基)氨基丙基甲基二甲氧基硅烷。
在步骤(b)中,还可以在作为聚合物链终端(M单元)组分的具有通式(3)或(4)的硅化合物的存在下进行环状硅氧烷的开环聚合:
R16R17R18Si(X) (3)
R16R17R18SiOSiR16R17R18 (4)
其中,R16、R17和R18各自独立地为氢或选自以下的基团:C1-C20烷基、C2-C20烯基、C6-C20芳基和具有至多50个硅原子的(聚)二甲基甲硅烷氧基,它们可以被(甲基)丙烯酰基、环氧乙烷基或氟基团取代,X为选自烷氧基、烯丙基、乙酰氧基、烯醇和氯的基团。所述硅化合物的量为至多100重量份,按照每100重量份的环状硅氧烷计。其后,蒸馏除去分散介质,生成无机颗粒-聚硅氧烷复合体。聚合物链终端组分的共存能够将各种官能团在端部引入至聚硅氧烷链上,这使得储存稳定性上的改进、与分散介质的相容性上的改进、粘度控制或引入反应性端基成为可能。
所添加的M单元组分的量优选为至多100重量份,更优选至多50重量份,按照每100重量份的环状硅氧烷计。大于100重量份的M单元组分是不期望的,因为可能阻碍聚硅氧烷的生长并且分散能力可能不足。当添加M单元组分时,所述量优选为至少0.01重量份,更优选至少0.1重量份,按照每100重量份的环状硅氧烷计。
具有式(3)的硅烷化合物的实例包括烷氧基硅烷,如三甲基甲氧基硅烷、三甲基乙氧基硅烷、三乙基甲氧基硅烷、正丙基二甲基甲氧基硅烷、正丙基二乙基甲氧基硅烷、异丙基二甲基甲氧基硅烷、异丙基二乙基甲氧基硅烷、丙基二甲基乙氧基硅烷、正丁基二甲基甲氧基硅烷、正丁基二甲基乙氧基硅烷、正己基二甲基甲氧基硅烷、正己基二甲基乙氧基硅烷、正戊基二甲基甲氧基硅烷、正戊基二甲基乙氧基硅烷、正己基二甲基甲氧基硅烷、正己基二甲基乙氧基硅烷、正癸基二甲基甲氧基硅烷和正癸基二甲基乙氧基硅烷;硅烷醇,如三甲基硅烷醇、三乙基硅烷醇、正丙基二甲基硅烷醇、正丙基二乙基硅烷醇、异丙基二甲基硅烷醇、异丙基二乙基硅烷醇、丙基二甲基硅烷醇、正丁基二甲基硅烷醇、正己基二甲基硅烷醇、正戊基二甲基硅烷醇和正癸基二甲基硅烷醇;烯丙基硅烷,如烯丙基三甲基硅烷、烯丙基三乙基硅烷和烯丙基三异丙基硅烷;乙酰氧基硅烷,如乙酰氧基三甲基硅烷、乙酰氧基三乙基硅烷、乙酰氧基三丙基硅烷和乙酰氧基三苯基硅烷;氯硅烷,如氯三甲基硅烷、氯三乙基硅烷、氯三丙基硅烷和氯三苯基硅烷;和烯醇硅烷,如异丙烯基氧基三甲基硅烷、三乙基异丙烯基氧基硅烷、异丙烯基氧基三丙基硅烷和三苯基异丙烯基氧基硅烷。
具有式(4)的硅氧烷化合物的实例包括六甲基二硅氧烷、六乙基二硅氧烷、六苯基二硅氧烷、1,1,3,3-四甲基二硅氧烷、1,3-二乙炔基-1,1,3,3-四甲基二硅氧烷、1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷、1,1,3,3-四苯基-1,3-二乙烯基二硅氧烷、1,3-双(巯基丙基)四甲基二硅氧烷、1,3-双(氨基丙基)四甲基二硅氧烷、1,3-双(乙酰氧基丙基)四甲基二硅氧烷和1,3-双(丙烯酰氧基丙基)四甲基二硅氧烷。
步骤(c)
所述方法可以进一步包括从由步骤(b)得到的无机颗粒-聚硅氧烷复合物的分散体中除去作为分散介质的极性有机溶剂的步骤(c)。分散介质的除去可以通过合适的手段如蒸馏或经由超滤的溶剂置换来进行。当通过蒸馏除去分散介质时,并行于分散介质的除去,加热促进聚硅氧烷的分子量增加,由此可以获得用较高分子量的聚硅氧烷涂覆的无机颗粒-聚硅氧烷复合物。
通过蒸馏除去分散介质优选在200至760mmHg,更优选300至760mmHg,且甚至更优选400至760mmHg的压力下进行。在低于200mmHg的压力下,操作可能不期望地因为混合物的暴沸而难于控制。高于760mmHg的压力可能阻止极性有机溶剂蒸发。
优选在50至250℃,更优选60至200℃,且甚至更优选80至150℃的温度进行蒸馏。低于50℃的温度是不期望的,因为蒸馏可能花费时间,而在高于250℃的温度,有机溶胶可能变化。调节压力,使得可以在所述范围的温度进行蒸馏。
蒸馏所需的热可以通过合适的加热手段如与加热介质热交换、感应加热或微波加热来提供。
步骤(d)
在所述方法中,可以在步骤(b)或(c)之后包括步骤(d):添加式(1)的环状硅氧烷、式(2)的聚合物扩链组分和/或式(3)或(4)的聚合物链终端组分并再次进行反应。通过增加步骤(d),可以将甚至在步骤(b)之前未溶于极性溶剂的化合物引入聚硅氧烷接枝结构部分,这使得精细控制物理性质包括折射控制、粘度控制、储存稳定性上的改进、与分散介质的相容性上的改进、分散性上的改进、以及引入反应性末端或反应性侧链成为可能。
实施例
通过阐释并且不是通过限制的方式在下文中给出本发明的实施例。所有份数为重量份。在25℃通过Ostwald粘度计测量粘度。
实施例1
合成具有氧化钛-氧化锡复合氧化物的核和氧化硅的壳的核/壳型颗粒的水分散体(TW-1)
将1.8g的氯化锡(IV)五水合物(Wako Pure Chemical Industries,Ltd.)添加至66.0g的36重量%氯化钛(IV)水溶液(商品名TC-36,由Ishihara Sangyo Kaisha,Ltd.制造),充分混合,并用1,000g去离子水稀释。将300g的5重量%氨水(Wako Pure Chemical Industries,Ltd.)逐渐添加至金属盐水溶液混合物,以进行中和和水解,由此使含锡的氢氧化钛沉淀。在此时,氢氧化钛浆料为pH 8。通过重复添加去离子水和倾析将氢氧化钛沉淀物去离子。向在去离子处理之后的含锡的氢氧化钛沉淀物中逐渐添加100g的30重量%过氧化氢水溶液(Wako Pure Chemical Industries,Ltd.)。随后在60℃搅拌3小时以充分反应。其后,添加纯水用于浓度调节,生成含锡的过钛酸的半透明溶液(固体浓度:1重量%)。向500-mL体积的高压釜(商品名TEM-D500,由Taiatsu Techno Corp.制造)装入350mL合成的过钛酸溶液,使其在200℃和1.5MPa的条件下经受240分钟的水热处理。其后,将高压釜中的反应混合物通过取样管转移至保持在25℃的水浴中的容器。通过该快速冷却猝灭反应,获得复合氧化物分散体(i)。
向配备有磁力搅拌器和温度计的可拆卸烧瓶中在室温(25℃)装入1,000份的复合氧化物分散体(i)、100份乙醇和2.0份氨,然后磁力搅拌。将可拆卸烧瓶置于冰浴中并冷却直至内容物的温度达到5℃。将18份四乙氧基硅烷(商品名KBE-04,由Shin-Etsu Chemical Co.,Ltd.制造)添加至可拆卸烧瓶,所述可拆卸烧瓶架置在μReactor EX(Shikoku Instrumentation Co.,Inc.)中,其中施加频率为2.45GHz和功率为1,000W的微波1分钟,同时继续磁力搅拌。在微波加热步骤期间监测温度计,确认内容物的温度达到85℃。将混合物通过定性过滤器(Advantec 2B)过滤,获得稀释的胶体分散体。通过经Dynafilter(商品名DyF152/S,由Mitsubishi Kakoki Kaisha,Ltd.制造,具有7nm平均孔径的MgAl2O盘(产品编号2065181,型号□152/7nm,由ANDRITZ KMPT GmbH制造))超滤,将稀释的胶体分散体浓缩至14重量%的固体浓度,生成由氧化钛-氧化锡复合氧化物的核和氧化硅的壳组成的核/壳型颗粒的水分散体(TW-1)。
合成核/壳型颗粒乙醇分散体(TE-1)
向配备有Dimroth冷凝器、氮气导入管、温度计和搅拌桨的2-L四颈可拆卸烧瓶装入300g(固体浓度14重量%)的无机颗粒分散体形式的核/壳型颗粒水分散体(TW-1)和3g作为催化剂的磺酸型阳离子交换树脂。向烧瓶中添加225g甲基三甲氧基硅烷(KBM-13,由Shin-Etsu Chemical Co.,Ltd.制造)并以250rpm强力搅拌。观察到在搅拌时分散体与甲基三甲氧基硅烷反应并且变得均匀和分散体的温度从25℃升至52℃。将分散体加热并在50℃的温度搅拌2小时,其后将其通过添加750g乙醇稀释,同时在250rpm搅拌。将经稀释的分散体引入具有陶瓷过滤装置的超滤仪器,在其中使用乙醇进行洗涤和溶剂置换直至滤出液达800g。将分散体从过滤室中取出。获得核/壳型颗粒乙醇分散体(TE-1),其具有17重量%的固体浓度,1.1重量%的水浓度和3nm的颗粒尺寸。所述颗粒尺寸为通过动态光散射法,特别是Nanotrac UPA-EX150(Nikkiso Co.,Ltd.)测量的体积基准颗粒尺寸分布中的50%累积颗粒尺寸(D50)。
合成聚硅氧烷接枝的无机氧化物(TP-1)
向配备有Dimroth冷凝器、氮气引入管、温度计和搅拌桨的2-L四颈可拆卸烧瓶装入258g具有17重量%的固体浓度的核/壳型颗粒乙醇分散体(TE-1)、634g乙醇、179g作为环状硅氧烷的八甲基环四硅氧烷(D4)和2g作为催化剂的三氟甲磺酸。将内容物在室温搅拌12小时并回流2小时,以进行开环聚合。在大气压下馏除乙醇,其后将反应混合物在110℃搅拌另外2小时。将混合物冷却至室温,此时向其中添加300g甲苯和8g合成水滑石(商品名Kyoward 500,Kyowa Chemical Industry Co.,Ltd.)。通过过滤除去其上吸附有酸催化剂的水滑石,其后在真空下馏除甲苯,生成液体聚硅氧烷接枝的无机氧化物(TP-1),即聚硅氧烷接枝至颗粒表面的无机氧化物颗粒-聚硅氧烷复合物。在热重分析(TGA)时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为72重量%。分析结果汇总于表1中。
实施例2
合成聚硅氧烷接枝的无机氧化物(TP-2)
通过与实施例1的TP-1合成中相同的工序制备液体聚硅氧烷接枝的无机氧化物(TP-2),不同之处在于使用12g的TE-1、28g乙醇、18g的D4和0.2g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为88重量%。分析结果汇总于表1中。
实施例3
合成聚硅氧烷接枝的无机氧化物(TP-3)
通过与实施例1的TP-1合成中相同的工序制备液体聚硅氧烷接枝的无机氧化物(TP-3),不同之处在于使用17g的TE-1、42g乙醇、7g的D4和0.2g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为64重量%。分析结果汇总于表1中。
实施例4
合成氧化铈乙醇分散体(CE-1)
通过与实施例1的TE-1合成中相同的工序制备氧化铈乙醇分散体(CE-1),不同之处在于使用150g具有20重量%固体浓度的氧化铈水分散体(NanoUse CE-T20B,由Nissan Chemical Industries,Ltd.制造)和150g去离子水的混合物替代TW-1。分散体(CE-1)具有15重量%的固体浓度,0.4重量%的水浓度和3nm的颗粒尺寸。
合成聚硅氧烷接枝的氧化铈(CP-1)
通过与实施例1中相同的工序制备液体聚硅氧烷接枝的氧化铈(CP-1),不同之处在于使用13g具有15重量%固体浓度的氧化铈乙醇分散体(CE-1)替代TE-1和使用27g乙醇、18g的D4和0.18g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为90重量%。分析结果汇总于表1中。
实施例5
合成聚硅氧烷接枝的氧化硅(SP-1)
通过与实施例1中相同的工序制备液体聚硅氧烷接枝的氧化硅(SP-1),不同之处在于使用100g具有30重量%固体浓度和45nm的颗粒尺寸的氧化硅IPA分散体(IPA-ST-L,由Nissan Chemical Industries,Ltd.制造)替代TE-1和使用500g乙醇、270g的D4和0.3g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为80重量%。分析结果汇总于表1中。
实施例6
合成聚硅氧烷接枝的无机氧化物(TP-4)
向配备搅拌器的100-mL单颈茄形烧瓶装入5g在实施例2中制备的TP-2、5g的D4和0.05g作为催化剂的三氟甲磺酸。将内容物在50℃搅拌3小时。将反应混合物冷却至室温,此时向其中添加30g甲苯和0.2g合成水滑石(Kyoward 500)。通过过滤除去其上吸附有酸催化剂的水滑石,其后在真空下馏除甲苯,生成液体聚硅氧烷接枝的无机氧化物(TP-4)。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为94重量%。分析结果汇总于表1中。
实施例7
合成聚硅氧烷接枝的无机氧化物(TP-5)
通过与实施例6中相同的工序制备聚硅氧烷接枝的无机氧化物(TP-5),不同之处在于使用1g在实施例2中制备的TP-2、9g的D4和0.09g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为97重量%。分析结果汇总于表1中。
实施例8
合成聚硅氧烷接枝的氧化铈(CP-2)
通过与实施例6中相同的工序制备聚硅氧烷接枝的氧化铈(CP-2),不同之处在于使用2g的CP-1替代TP-2和使用18g的D4和0.18g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为98重量%。分析结果汇总于表1中。
实施例9
合成聚硅氧烷接枝的氧化硅(SP-2)
通过与实施例6中相同的工序制备聚硅氧烷接枝的氧化硅(SP-2),不同之处在于使用2g的SP-1替代TP-2,和使用18g的D4和0.18g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为98重量%。分析结果汇总于表2中。
实施例10
合成聚硅氧烷接枝的无机氧化物(TP-6)
向具有搅拌器的100-mL单颈茄形烧瓶装入18g在实施例1中制备的TP-1、2g作为M单元的六甲基二硅氧烷、1g的H2O和0.02g作为催化剂的三氟甲磺酸。将内容物在110℃搅拌6小时。向反应混合物中添加30g甲苯、1g硫酸钠和0.08g水滑石(Kyoward 500)用于脱水和酸催化剂吸附。通过过滤除去固体,其后在真空下馏除甲苯,生成聚硅氧烷接枝的无机氧化物(TP-6)。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为70重量%。分析结果汇总于表2中。
实施例11
合成聚硅氧烷接枝的无机氧化物(TP-7)
通过与实施例1的TP-1合成中相同的工序制备液体聚硅氧烷接枝的无机氧化物(TP-7),不同之处在于使用12g的TE-1、28g乙醇、12g的D4、2g六甲基二硅氧烷、5g二甲氧基二苯基硅烷(KBM-202,由Shin-Etsu Chemical Co.,Ltd.制造)和0.2g三氟甲磺酸。在TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为89重量%。分析结果汇总于表2中。
对比例1
从实施例1中在环状硅氧烷处理之前的核/壳型颗粒乙醇分散体(TE-1)中,在真空下馏除分散介质,留下颗粒样品,将其通过TGA分析。在环状硅氧烷处理之前在全部颗粒中的硅氧烷成分含量为9重量%。将乙醇分散体(TE-1)用乙醇稀释,以形成具有5重量%的固体浓度的另一样品,评价其分散性。结果汇总于表2中。
对比例2
从实施例4中在环状硅氧烷处理之前的氧化铈乙醇分散体(CE-1)中,在真空下馏除分散介质,留下颗粒样品,将其通过TGA分析。在环状硅氧烷处理之前在全部颗粒中的硅氧烷成分含量为8重量%。将乙醇分散体(CE-1)用乙醇稀释,以形成具有5重量%的固体浓度的另一样品,评价其分散性。结果汇总于表2中。
对比例3
从具有30重量%的固体浓度的氧化硅异丙醇分散体(IPA-ST-L,Nissan Chemical Industries,Ltd.)中,在真空下馏除分散介质,留下样品,将其通过TGA分析。固体中的氧化硅颗粒含量为98重量%。将分散体IPA-ST-L用乙醇稀释以形成具有5重量%的固体浓度的另一样品,评价其分散性。结果汇总于表2中。
对比例4
向50g核/壳型颗粒乙醇分散体(TE-1)中添加8.5g(等于无机颗粒中的固体重量)的一端用三甲氧基甲硅烷氧基封端的二甲基聚硅氧烷(X-24-9822,由Shin-Etsu Chemical Co.,Ltd.制造)作为在一端具有反应性三甲氧基甲硅烷氧基的聚硅氧烷(Q单元)和0.05g磺酸型阳离子交换树脂。将混合物回流6小时。在真空下从产生的样品中馏除分散介质并通过过滤收集沉淀的固体。在沉淀的固体的TGA时,全部无机颗粒-聚硅氧烷复合物中的硅氧烷成分的含量为12重量%。基于该结果和对比例1中的颗粒核中的硅氧烷成分的重量损失,由D单元和M单元组成的接枝成分的含量经计算为3重量%。结果汇总于表2中。
[评价无机颗粒-聚硅氧烷复合物]
通过以下仪器和方法评价实施例1至11和对比例1至4中的样品。
无机颗粒和聚硅氧烷成分的含量
通过将在TGA之后的残余物的重量视为样品中的无机颗粒的重量,和将重量损失视为聚硅氧烷成分的重量测定无机颗粒和聚硅氧烷成分的含量。通过Thermo Plus系统(Rigaku Corp.)进行TGA。在25℃至900℃的温度范围内使用铂盘在氮气气氛中进行分析。
ΣD/ΣM值
将氘代氯仿添加至样品,从而提供40重量%样品溶液。将几滴预先制备的1M的乙酰丙酮化铬(III)在氘代氯仿(二者均由Kanto Chemical Co.,Ltd.制造)中的溶液添加至样品溶液,将其转移至直径10mm的PTFE的NMR管,并通过29Si-NMR光谱分析。分析条件包括门控去耦、45°脉冲和6秒脉冲间隔的脉冲序列、11.75T的磁场强度和2,220次扫描数。从分析结果计算归属于全部D单元的29Si-NMR信号的积分值(ΣD)和归属于全部M单元的29Si-NMR信号的积分值(ΣM),并确定它们的比例ΣD/ΣM。
分散性
将每个样品以10重量%的浓度分散于硅油中。将分散液通过三脚架悬挂式离心机(H-100F,由KOKUSAN Co.,Ltd.制造)在2000(×g)的相对离心力离心60分钟,其后观察无机颗粒的沉淀或相分离。关于分散性,当在具有50mm2/s粘度的硅油(KF-96-50CS,由Shin-Etsu Chemical Co.,Ltd.制造)中观察到沉淀时将样品分级为差“×”,当在相同的硅油中观察不到沉淀时分级为好“○”,当即使在具有10,000mm2/s粘度的粘性硅油(KF-96H-10000CS,Shin-Etsu Chemical Co.,Ltd.)中也观察不到沉淀时分级为非常好“◎”。
表1
表2
注释:
(1)在对比例1至3中,通过在真空蒸馏分散介质之后留下的无机颗粒固体的TGA测定颗粒含量。
(2)在对比例4中,通过在真空蒸馏分散介质之后留下的无机颗粒固体和通过从在真空蒸馏分散介质之后留下的清澈液体过滤收集的无机颗粒的TGA测定颗粒含量。
(3)在对比例4中,通过充当反应物的在一端用三甲氧基甲硅烷氧基封端的二甲基聚硅氧烷(X-24-9822,由Shin-Etsu Chemical Co.,Ltd.制造)的29Si-NMR测量硅氧烷成分的ΣD/ΣM值。
在实施例1至5中,通过以下合成无机颗粒-聚硅氧烷复合物:制备由作为分散质的无机颗粒和作为分散介质的乙醇和/或IPA组成的有机溶胶,在分散体中进行环状硅氧烷的开环聚合,馏除分散介质。在对比例1至3中,可以将未用环状硅氧烷处理的样品(TE-1、CE-1和IPA-ST-L)分散于极性溶剂如醇中,不可以分散于非极性溶剂如己烷和硅油中,即它们聚集和沉淀。相比之下,实施例1至5的无机颗粒-聚硅氧烷复合物显示出在己烷和硅油中的高分散性。推测颗粒显示出这样的高分散性是因为它们由于如下的机理而变得高度疏水:环状硅氧烷的开环聚合和无机颗粒表面上的羟基或烷氧基与聚硅氧烷端部之间的反应同时发生,由此将聚硅氧烷接枝至无机颗粒表面。
在对比例4中,其中颗粒具有接枝至它们的表面的一端用三甲氧基甲硅烷氧基封端的二甲基聚硅氧烷(X-24-9822,Shin-Etsu Chemical Co.,Ltd.),尽管聚硅氧烷成分的ΣD/ΣM值大于实施例1至5,即链长更长,但是颗粒并不显示出在己烷和硅油中的分散性。如表2中所示,在对比例4中,聚硅氧烷的被覆量(coverage)小于20份,按每100份无机颗粒计。这表明这样的低的被覆量不足以避免无机颗粒之间的直接接触。此外,高分子量的聚硅氧烷难于以高密度接枝至颗粒表面,因为高分子量的聚硅氧烷在分子末端反应性差。
在实施例1至5中,颗粒具有以比现有技术更高的密度接枝至其上的聚硅氧烷,据推测因为较高反应性的低分子量硅氧烷接枝至颗粒表面,然后在颗粒表面上发生聚合物生长反应。在实施例6至9中,使在实施例1至5中制备的无机颗粒-聚硅氧烷复合物进一步与环状硅氧烷反应,生成具有其上接枝有较高分子量的聚硅氧烷的复合物。所产生的复合物可充分分散于一般的无机颗粒几乎不会分散于其中的非常粘稠的硅油(粘度为5,000至10,000mm2/s)中。据推测这是因为无机颗粒由于接枝至其上的高分子量聚硅氧烷变得高度疏水。
由表1和2的结果可知,随着引入的环状硅氧烷的量变得更大,聚硅氧烷的ΣD/ΣM值变得更大,尽管没有引入聚合物链终端组分(M单元)。典型地,在不存在任何终端组分下的开环聚合产生具有显著高的分子量的胶状(gum-like)聚硅氧烷。因此暗示在实施例中,颗粒表面充当聚合物链终端组分起作用,即聚硅氧烷在颗粒表面上生长。
在实施例10中,使实施例1中制备的无机颗粒-聚硅氧烷复合物与六甲基二硅氧烷反应用于聚合物末端改性。图1显示了在实施例1和10中制备的复合物的1H-NMR光谱。从这些NMR图看出,归属于末端乙氧基的峰在处理之后明显减少,表明末端改性的进行。
已证实的是,本发明的无机颗粒-聚硅氧烷复合物可以通过在普通反应器中使用多用途的环状硅氧烷化合物作为反应物并且进行聚硅氧烷的合成和聚硅氧烷于颗粒表面的接枝而制备。可以将无机颗粒-聚硅氧烷复合物以任意期望的浓度分散至多种硅油和有机溶剂中,并且此外甚至在普通的无机颗粒几乎不会分散于其中的高度粘稠的硅油中也均匀地分散。如典型的有机硅流体那样,所述复合物具有通过增加分子量、引入交联点和进行分子改性如末端改性而可调节的粘性、分散性和物理性质。因为在此以前从未报道特征在于容易用官能团改性和在非极性溶剂中的高分散性的无机颗粒材料,所以认为本发明是工业上独特的和有用的。
实施例12
将1g实施例7中制备的聚硅氧烷接枝的无机氧化物(TP-5)、10g在末端取代有乙烯基二甲基甲硅烷氧基的聚二甲基硅氧烷油(粘度为5,000mm2/s)、0.4g作为交联剂的有机氢聚硅氧烷(KE1800E,由Shin-Etsu Chemical Co.,Ltd.制造)和0.1g氢化硅烷化催化剂(X-93-405,由Shin-Etsu Chemical Co.,Ltd.制造)的混合物脱气并且浇铸至经涂覆的6cm×3cm×0.1cm的模具中,在所述模具中使其在150℃加成固化15分钟,生成在其中分散有无机颗粒-聚硅氧烷复合物的透明硅橡胶。结果汇总于表3中。
实施例13
重复与实施例12中相同的工序,不同之处在于使用1g实施例8中制备的聚硅氧烷接枝的氧化铈细颗粒(CP-2)替代实施例12中的TP-5。获得其中分散有氧化铈-聚硅氧烷复合物的透明硅橡胶。结果汇总于表3中。
实施例14
重复与实施例12中相同的工序,不同之处在于使用1g实施例9中制备的聚硅氧烷接枝的氧化硅细颗粒(SP-2)替代实施例12中的TP-5。获得其中分散有氧化硅-聚硅氧烷复合物的透明硅橡胶。结果汇总于表3中。
对比例5
重复与实施例12中相同的工序,不同之处在于使用0.2g实施例1中制备的核/壳型颗粒乙醇分散体(TE-1)替代实施例12中的TP-5。获得包含无机颗粒的硅橡胶。结果汇总于表3中。
对比例6
重复与实施例12中相同的工序,不同之处在于使用0.2g实施例4中制备的氧化铈乙醇分散体(CE-1)替代实施例12中的TP-5。获得包含氧化铈的硅橡胶。结果汇总于表3中。
对比例7
重复与实施例12中相同的工序,不同之处在于使用0.03g氧化硅IPA分散体(IPA-ST-L,由Nissan Chemical Industries,Ltd.制造)替代实施例12中的TP-5。获得包含氧化硅的硅橡胶。结果汇总于表3中。
对比例8
将10g在末端取代有乙烯基二甲基甲硅烷氧基的聚二甲基硅氧烷油(粘度为5,000mm2/s)、0.4g交联剂(KE1800E,由Shin-Etsu Chemical Co.,Ltd.制造)和0.1g氢化硅烷化催化剂(X-93-405,由Shin-Etsu Chemical Co.,Ltd.制造)的混合物脱气并且浇铸至经涂覆的6cm×3cm×0.1cm的模具中,在所述模具中使其在150℃加成固化15分钟,生成不包含无机颗粒的透明硅橡胶。结果汇总于表3中。
[评价包含无机颗粒-聚硅氧烷复合物的硅橡胶]
通过以下仪器和方法评价实施例12至14和对比例5至8中的样品。
颗粒含量
通过将在TGA之后的残余物的重量视为无机颗粒的重量和将重量损失视为聚硅氧烷成分的重量,计算颗粒含量。通过Thermo Plus系统(Rigaku Corp.)进行TGA。在25℃至900℃的温度范围内使用铂盘在氮气气氛中进行分析。
雾度测量
通过雾度计(NDH2000,Nippon Denshoku Industries Co.,Ltd.)测量1mm厚的膜样品的雾度。
可见光透射率
通过分光光度计U-3900H(Hitachi High-Tech Science Corp.)测量1mm厚的膜样品的透射率。将在500至700nm波长范围内透射率为至少80的样品分级为好“○”并将在500至700nm波长范围内透射率为小于80的样品分级为差“×”。
表3
在对比例8中制备不包含无机颗粒的硅橡胶,而在实施例12至14和对比例5至7中制备包含无机颗粒的硅橡胶。在对比例5至7中的没有用环状硅氧烷处理的无机颗粒的分散体中,颗粒在硅油中聚集,导致光散射的增加、透明度的降低和雾度的显著增加。另一方面,包含如在实施例12至14中的用环状硅氧烷处理的无机颗粒-聚硅氧烷复合物的硅橡胶具有如下优点:光散射受到抑制,因为颗粒充分分散于硅油中,并且透明度和雾度值处于与对比例8中不包含无机颗粒的硅橡胶相同的水平。
从图2至4中的UV/Vis透射光谱看出,对比例5至7在500至700nm的可见光范围内由于白色混浊而具有显著降低的透射率,而实施例12至14保持高透明度,即相比于对比例8中的不含无机颗粒的样品在可见范围内基本上没有透射率的降低。在包含UV吸收性无机颗粒如氧化钛和氧化铈的对比例5和6中,接近300nm的光吸收弱。相比之下,包含等当量的量的相同无机颗粒的实施例12和13的样品在300nm以上的光吸收得到急剧地改进。这可能归因于以下机理。对比例5和6的样品中的颗粒由于聚集而具有明显降低的表面积,导致光吸收效率的明显下降。在实施例12和13的样品中的颗粒充分分散,保持表面积基本上不变并且有效地吸收光。
如上文所描述,根据本发明获得的硅橡胶具有充分分散在有机硅中的无机颗粒,使得其可以保持透明度并且相比于常规材料还可以更有效地开发有机硅材料中的无机颗粒的UV吸收能力和其它功能。本发明在工业上也是有用的并且预期应用于许多领域。
出于阐释本发明的有利之处的目的提出这些实施例并且这些实施例并不意在限制本发明的范围。
工业应用性
本发明的无机颗粒-聚硅氧烷复合物可使无机颗粒充分分散在有机硅材料和不包含极性基团的各种有机材料中。因此,可以将无机颗粒-聚硅氧烷复合物应用至涂料组合物、发光元件封装、导热组合物、耐热组合物、UV吸收性组合物等,且特别适合于要求高透明度、耐候性和导热性的领域。尽管已将各种机械技术用于这些领域以均匀地分散无极氧化物颗粒,但是使用与粘结剂完全相容的无机颗粒-聚硅氧烷复合物不仅有助于技术的简化,而且使得颗粒的充分分散成为可能,使得颗粒更有效地发挥它们的功能。因此,本发明扩展了在制备组合物时的材料的选择并且有助于本领域的工业进步。
通过引用将日本专利申请第2015-239303号并入本文中。
尽管已对一些优选的实施方案进行了描述,但根据上述教导可对其进行许多变形和改变。因此可理解,在不脱离所附权利要求的范围的情况下可在具体描述以外实施本发明。
- 还没有人留言评论。精彩留言会获得点赞!