重定向T细胞以治疗HIV感染的方法与流程
本申请以35u.s.c.§119(e)要求2015年9月22日提交的美国临时专利申请号62/222,132和2015年11月11日提交的美国临时专利申请号62/253,790的优先权,其通过引用以其全部并入本文。
关于联邦资助的研究或研发的声明
本发明在美国国立卫生研究给予的批准号ai104280、ai117950、和ar064220下使用政府支持完成。政府具有本发明中的某些权利。
背景技术
不管抗逆转录病毒疗法最小化人免疫缺陷病毒类型(hiv)复制并且增加患者寿命和生活质量的能力如何,与终身治疗方案相关联的健康结果和经济负担致使彻底治愈是极具吸引力的。虽然t细胞在控制病毒复制中发挥重要作用,但是它们自身是hiv介导的破坏的靶标。
cd4t细胞活性的恢复,不论通过免疫强化或通过防止缺失,是在不存在高活性抗逆转录病毒疗法(haart)的情况下使得能够长期控制hiv复制的关键因素。将t细胞制造为治疗剂以治疗hiv的尝试已经进行了超过二十年。t细胞可以被改造以表达称为嵌合抗原受体(car)的合成免疫受体,其由细胞外靶向抗体和细胞内信号传导结构域组成。此新的研究领域,称为过继性t细胞疗法,最近已经经历了许多技术进步。重要地,t-细胞疗法途径具有保护辅助性cd4t细胞和使它们装备直接抗病毒功能的潜力,其对在不存在抗逆转录病毒疗法的情况下改进hiv特异性细胞毒性和取得hiv复制的控制可以是至关重要的。虽然在用于过继性疗法的t细胞工程领域中已经取得重大进步,包括展现安全性和可行性,但是没有临床试验在不存在haart的情况下导致对hiv-复制的持久性和一致性控制。
与疫苗途径——其依赖于患者自己体内hiv特异性淋巴细胞的产生和引发——形成对比,过继性t-细胞疗法提供了在施用之前定制治疗性t细胞的机会。因而,不管使用t细胞的直接基因操纵的不成功的治疗性尝试如何,清楚的是过继性细胞疗法可以通过生成hiv-抗性细胞、重定向hiv特异性免疫应答、或两种策略的组合有助于功能性治愈。然而,目前,不清楚如何最好地改造t细胞,使得可以在不存在抗逆转录病毒疗法的情况下取得对hiv复制的持续控制。
本领域对抑制hiv复制和提供基因疗法介导的功能性治愈的更有效的t-细胞基因-工程和基因-编辑策略存在极大需要。本发明解决此需要。
技术实现要素:
如本文描述的,本发明涉及使用cd4膜结合嵌合受体或hiv特异性单链可变片段(scfv)car治疗感染hiv的哺乳动物的组合物和方法。
在一方面,本发明包括编码膜结合嵌合受体的分离的核酸序列。分离的核酸序列包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞。
在另一方面,本发明包括编码膜结合嵌合受体的分离的氨基酸序列。分离的氨基酸序列包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞。
在一些实施方式中,cd4细胞外结构域由包括seqidno:3或64的核酸序列编码。在一些实施方式中,cd4细胞外结构域包括氨基酸序列seqidno:46。在一些其它实施方式中,cd4细胞外结构域特异性地结合至hiv包膜(env)糖蛋白。
在一些实施方式中,跨膜结构域包括由核酸序列seqidno:4或65编码的cd8α铰链并且跨膜结构域包括由如下核酸序列编码的至少一种结构域:其包括选自seqidno:5和8的一种。在其它实施方式中,跨膜结构域包括seqidno:47的cd8α铰链,并且跨膜结构域包括选自seqidno:48和49的至少一种氨基酸序列。
在其它实施方式中,信号传导结构域包括由包括seqidno:6或68的核酸序列编码的cd3ζ信号传导结构域。在其它实施方式中,信号传导结构域包括包含seqidno:51的cd3ζ信号传导结构域。
在又其它实施方式中,本发明的分离的核酸或氨基酸序列进一步包括共刺激信号传导区。
在本文描绘的发明的上面的方面或任何其它方面的多种实施方式中,共刺激信号传导区包括选自tcrζ的α、β或ζ链,fcrγ,fcrβ,cd3γ,cd3δ,cd3ε,cd5,cd22,cd79a,cd79b,或cd66d的共刺激分子的细胞内结构域,来自cd27、cd28、4-1bb(cd137)、dap12、ox9、ox40、cd30、cd40、pd-1、icos、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、与cd83特异性地结合的配体的共刺激信号传导区,和其任意组合。在一些实施方式中,共刺激信号传导区是cd28并且由包括seqidno:9或67的核酸序列编码。在其它实施方式中,共刺激信号传导区是cd28并且包括seqidno:49。
在一方面,本发明包括载体,其包括编码膜结合嵌合受体的分离的核酸序列。载体的分离的核酸序列包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞。在一些实施方式中,载体的核酸序列包括选自seqidno:1、7、62和63的至少一种。
在另一方面,本发明包括载体,其包括膜结合嵌合受体。载体的膜结合嵌合受体包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞,其中载体包括选自seqidno:44和45的至少一种氨基酸序列。在一些实施方式中,载体包括efα启动子。
在又另一个方面,本发明包括编码嵌合抗原受体(car)的分离的核酸序列。编码本发明的car的分离的核酸序列包括hiv特异性结合结构域、跨膜结构域、共刺激信号传导区、和信号传导结构域,其中hiv结合结构域包括抗-hiv抗体或其片段。
在一些实施方式中,hiv特异性结合结构域包括重和轻链。在其它实施方式中,hiv特异性结合结构域是人抗体、人源化抗体、和其片段。在其它实施方式中,抗体或其片段选自fab片段、f(ab')2片段、fv片段、和单链fv(scfv)。在又其它实施方式中,scfv包括选自seqidno:13、17、21、25、29、57-60和61的氨基酸序列。在进一步的实施方式中,scfv由选自seqidno:12、16、20、24、28、75-78和79的核酸序列编码。在还进一步的实施方式中,hiv特异性结合结构域特异性地结合至感染hiv的细胞的表面,或hiv病毒粒子。
在一些实施方式中,本发明的car的共刺激信号传导区包括选自tcr、cd3ζ、cd3γ、cd3δ、cd3ε、cd86、普通的fcrγ、fcrβ(fcεr1b)、cd79a、cd79b、fcγriia、dap10、dap12、t细胞受体(tcr)、cd8、cd27、cd28、4-1bb(cd137)、ox9、ox40、cd30、cd40、pd-1、icos、kir家族蛋白、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、与cd83特异性地结合的配体、cds、icam-1、gitr、baffr、hvem(lightr)、slamf7、nkp80(klrf1)、cd127、cd160、cd19、cd4、cd8α、cd8β、il2rβ、il2rγ、il7rα、itga4、vla1、cd49a、itga4、ia4、cd49d、itga6、vla-6、cd49f、itgad、cd11d、itgae、cd103、itgal、cd11a、lfa-1、itgam、cd11b、itgax、cd11c、itgb1、cd29、itgb2、cd18、lfa-1、itgb7、tnfr2、trance/rankl、dnam1(cd226)、slamf4(cd244、2b4)、cd84、cd96(tactile)、ceacam1、crtam、ly9(cd229)、cd160(by55)、psgl1、cd100(sema4d)、cd69、slamf6(ntb-a、ly108)、slam(slamf1、cd150、ipo-3)、blame(slamf8)、selplg(cd162)、ltbr、lat、gads、slp-76、pag/cbp、nkp44、nkp30、nkp46、nkg2d、toll样受体1(tlr1)、tlr2、tlr3、tlr4、tlr5、tlr6、tlr7、tlr8、tlr9、和其任意组合的共刺激分子的细胞内结构域。
在一些实施方式中,本发明的car的信号传导结构域包括由核酸序列seqidno:6或68编码的cd3ζ信号传导结构域。
在一些实施方式中,car由选自seqidno:10、14、18、22、26、34、70-73、和74的核酸序列编码。在其它实施方式中,car具有选自seqidno:11、15、19、23、27、35、52-55、和56的氨基酸序列。
在一方面,本发明包括修饰的细胞,其包括分离的核酸序列,该分离的核酸序列包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞;或分离的核酸序列,该分离的核酸序列编码包括hiv特异性结合结构域、跨膜结构域、共刺激信号传导区、和信号传导结构域的car,其中hiv结合结构域包括抗-hiv抗体或其片段。
在一些实施方式中,本发明细胞的修饰的细胞选自t细胞、自然杀伤(nk)细胞、细胞毒性t淋巴细胞(ctl)、和调节性t细胞。在其它实施方式中,修饰的细胞的核酸序列选自dna和mrna。在又其它实施方式中,核酸序列通过选自电穿孔、使用慢病毒、使用逆转录病毒和基于化学的转染的至少一种程序被引入细胞。
在另一方面,本发明包括组合物,其包括如本文上面列出的本发明的修饰的细胞。
在另一方面,本发明包括本发明的修饰的细胞在制造用于治疗需要其的对象中hiv感染的药物中的用途。
在又另一个方面,本发明包括药物组合物,其包括本发明的修饰的细胞和药学上可接受的运载体。
在进一步的方面,本发明包括用于刺激感染hiv的哺乳动物中细胞免疫应答的方法。方法包括给哺乳动物施用有效量的如本文上面列出的本发明的修饰的细胞。
在又进一步的方面,本发明包括治疗感染hiv的哺乳动物的方法。方法包括给哺乳动物施用如本文上面列出的本发明的修饰的细胞。在一些实施方式中,修饰的细胞对哺乳动物是自体的。在其它实施方式中,本发明的方法进一步包括给哺乳动物施用抗逆转录病毒疗法(haart)。在又其它实施方式中,修饰的细胞和haart被共同施用至哺乳动物。
附图说明
当连同附图阅读时,将更好地理解本发明的优选实施方式的下列详细描述。出于说明本发明的目的,在附图中显示了当前优选的实施方式。然而,应当理解本发明不限于在附图中显示的实施方式的精确的布置和工具。
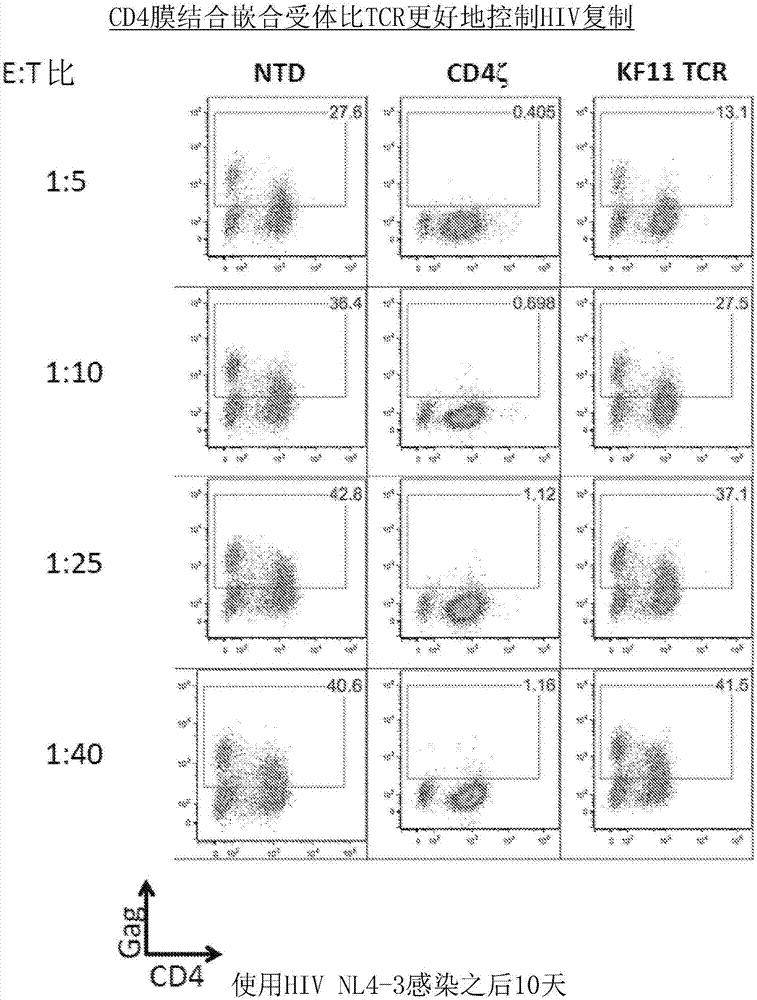
图1a-1b是展现cd4car在体外比hiv特异性精英控制者(elitecontroller)tcr强力超过100-倍的系列图。图1a:cd4和cd8t细胞获得自hla-b57+正常供体。以hivbal感染cd4t细胞,并且24小时之后,未转导的cd8t细胞(ntd)、表达特异于kafspevipmf-b57-kf11(kf11)的hla-b57局限的tcr的cd8t细胞、或表达ef1α-cd8αtmcd4car构建体(cd4z)的cd8t细胞,后二者均在ef1α启动子下表达,以指示的效应物/靶标比混合。转导效率在共培养之前被归一化至40%。在6天的共培养后,针对cd8阴性t细胞显示gagp24和cd4染色。cd4膜结合嵌合受体(还称为cd4ζ或cd4ζ构建体)比hla-b*57局限的kf11精英控制者tcr更好地控制nl4-3hiv-1复制。kf11tcr与患者中对hiv-1复制的较好控制相关联并且是针对hiv-1的最强力的患者源tcr之一。这代表使用基于tcr的策略可以取得的对hiv-1的最佳控制。与kf11tcr形成对比,cd4ζ能够在显示的所有效应细胞/靶标细胞(e:t)比下完全地控制hiv-1复制。图1b:在cd8阴性细胞上门控的以一式三份进行的单个实验的汇总数据。误差棒指示平均值的标准误差(sem)。这些数据代表3个独立实验。
图2a-2g是描绘启动子和跨膜改变改进cd4膜结合嵌合受体功效和增加对hiv-1的控制的系列示意图示和图。图2a:与包含pgk启动子和cd4tm结构域的进入临床试验的构建体(底)比较,具有cd8α跨膜(tm)和ef1α启动子的新的、改进的cd4膜结合嵌合受体构建体(顶)的示意图。图2b:转导的cd8中cd4ζ的表面表达。与进入临床试验的基于逆转录病毒载体的构建体(最右)比较,当使用慢病毒载体时,以更高的水平表达相同的构建体(左二)。当ef1α启动子被置换入慢病毒载体时,此表达被进一步增加(左二)。图2c:以演示显示的所有数据的实验时间线。使用αcd3/cd28涂覆的珠和100-300iu/mlil-2刺激健康供体cd4和cd8t淋巴细胞。在24小时后,使用慢病毒转导cd8,或在第3和5天使用逆转录病毒进行转导。在5天后,去除珠,并且在48小时之后感染cd4t细胞。在感染cd4t细胞后24小时,以不同的e:t比共培养cd8。培养物每隔一天进料培养基和il-2并且进行cd4、cd8、和细胞内p24染色直到hiv-1复制在ntd孔中达到最大复制能力(通常在第10-12天左右)。图2d-2e:以hiv-1bal感染cd4t细胞之后8天的细胞内p24染色。图2d,在cd4t细胞上门控,和图2e,在cd8t细胞上门控。具有慢病毒载体、ef1α启动子、和cd8α跨膜结构域的新的、改进的cd4ζ构建体在比具有pgk启动子和cd4跨膜结构域的构建体(在1:5和1:10下失控)低得多的e:t比下(直到大概1:100)控制hiv-1。虽然当与逆转录病毒pgkcd4tm构建体比较时观察到更高表达的慢病毒pgkcd4tm构建体,但是在hiv-1复制的控制方面可见小益处。重要地,较高表达的ef1αcd8tm构建体不促进较高的感染速率,这是因为cd8保持未感染直到大约1:100比率,而pgkcd4tm+cd8在较低的e:t比下变得感染。图2f:在cd8阴性细胞上门控的以一式三份进行的单个实验的汇总数据。误差棒指示平均值的标准误差(sem)。图2g:cd8阴性t细胞中的细胞内p24在实验的时间过程内的测量水平。每个图代表不同的e:t比。这些数据代表三个独立实验。
图3a-3e是系列示意图示和图,其描绘ef1α启动子和cd8α跨膜改变是改进car表达和hiv-1的控制的至关重要的修饰。图3a:四种慢病毒载体嵌合受体构建体——其被生成以比较改变启动子和跨膜组分每个如何各自地影响对hiv-1复制的控制——的示意图。顶部构建体指的是什么被用于临床试验但是现在被插入慢病毒载体。图3b:活化后8天cd8t细胞上的cd4car表达。原代人cd8t细胞使用αcd3/αcd28涂覆的珠被活化并且保持未转导的(ntd)或转导指示的慢病毒载体。在8天的培养后,通过cd4染色测量car表达。在每个图上指示每个构建体的中值荧光强度(mfi)。图3c:以hiv-1bal感染cd4t细胞之后8天的细胞内p24染色。左柱集在cd4t细胞上门控和右柱集在cd8t细胞上门控。如先前可见的,ef1αcd8tm构建体在最低e:t比下控制hiv-1。具有ef1α启动子(中柱)的cd4tm的单纯增加的表达改进了对hiv-1复制的控制,但是ef1α启动子和cd8α跨膜置换的增加的表达的组合产生对hiv-1的最佳控制。cd8αtm似乎减少以嵌合受体改造的cd8t细胞的感染(右柱集),特别是在1:50比率(ef1α构建体)和1:25比率(pgk构建体)下。图3d:在cd8阴性细胞上门控的以一式三份进行的单个实验的汇总数据。误差棒指示平均值的标准误差(sem)。图3e:cd8阴性t细胞中的细胞内p24在实验的时间过程内的水平。每个图代表不同的e:t比。这些数据代表三个独立实验。
图4a-4c是展现基于单链可变片段(scfv)广泛中和抗体(bnab)的car和cd4膜结合嵌合受体二者可以控制hiv-1感染的系列表格和图。图4a:在假设这些强力和广泛中和的hiv特异性抗体将提供靶向感染hiv的细胞的替代手段下生成由hiv-1特异性bnab改装的一组单链可变片段(scfv)car。以它们的hiv-1结合宽度和中和效力排列的一组scfv(参见图4a中的表格)被克隆入相同的ef1α-cd8αtm-ζ构建体骨架作为cd4膜结合嵌合受体,并且评估它们控制hiv-1复制的能力。在活化之后两周,这些t细胞与cr51标记的k562靶标细胞——其在指示的效应物/靶标比下表达hiv-1yu2gp160——混合。标绘靶标的特异性裂解。标绘的数据显示了三个独立实验的平均数。来自4小时共培养的cr51释放杀伤试验显示了cd8t细胞表达scfvcar或cd4膜结合嵌合受体中任一种,而非未转导的(ntd)t细胞、裂解的表达env的靶标细胞。这指示scfvcar以适当的折叠/构象表达并且能够结合env。图4b:原代人cd8t细胞使用αcd3/αcd28涂覆的珠被活化并且保持未转导的(ntd)或转导ef1α-cd8αtm慢病毒载体——其编码源自vrc01、3bnc60、pg9、pgt128、或pgdm1400抗体的hiv特异性car,或cd4car。活化之后两周,cd8t细胞在1:1比率下与表达hiv-1yu2gp160的k562细胞共培养6小时,并且测量细胞内ifnγ和mip-1β产生。转导效率在共培养之前被归一化至60%。图4c:使用在图2c中概述的实验设计,测试hiv特异性car在原代人cd4t细胞中控制hiv-1复制的能力。转导效率在共培养之前被归一化至70%。在以hiv-1bal感染cd4t细胞之后9天进行细胞内gagp24和cd4染色,显示cd4t细胞。cd4膜结合嵌合受体和scfvcar二者展现了相对于ntd对照高至1:50-1:200的e:t比的env靶标细胞,其优于在图1a中显示的kf11tcr的活性,该kf11tcr展现了高至1:25e:t比的功效。误差棒指示平均值的标准误差(sem)。数据代表三个独立实验。
图5a-5b是系列图,其显示cd28共刺激促进对hiv-1bal的控制并且cd28和4-1bb共刺激对体外hiv-1复制的控制具有相反作用。图5a:在以hiv-1bal感染cd4t细胞之后10天进行细胞内p24染色,显示cd4t细胞。已经用于普通car设计的一组共刺激结构域被克隆入cd4膜结合嵌合受体骨架以测定共刺激对hiv-1体外控制的作用。与cd4ζ构建体同样地或更好地一致控制hiv-1的唯一的膜结合嵌合受体是cd4cd28ζ构建体。相比之下,4-1bb、icos、和cd27的添加损害对hiv-1的控制,并且ox40和cd28-4-1bbζ组合似乎具有极小作用。图5b:在cd8阴性t细胞上门控的以一式三份进行的单个实验的汇总数据。误差棒指示平均值的标准误差(sem)。数据代表三个独立实验。
图6是cd4膜结合嵌合受体的核苷酸序列(cd4ζ构建体;seqidno:1)。此序列已经以红色注释与ef1α启动子相关的部分(seqidno:2),以黄色注释cd4细胞外结构域(seqidno:3),以绿色注释cd8α细胞外铰链(seqidno:4),以蓝绿色注释cd8α跨膜结构域(seqidno:5)和以粉色注释cd3ζ(seqidno:6)。
图7是cd4cd28膜结合嵌合受体的核苷酸序列(cd4cd28ζ构建体;seqidno:7)。此序列已经以红色注释与ef1α启动子相关的部分,以黄色注释cd4细胞外结构域,以绿色注释cd8α细胞外铰链,以蓝绿色注释cd28跨膜结构域(seqidno:8),以蓝色注释cd28共刺激结构域(seqidno:9)和以粉色注释cd3ζ。
图8是基于五种抗体的car构建体的核苷酸和氨基酸(aa)序列表:pgdm1400杀伤细胞免疫球蛋白样受体(kir)核苷酸和aa序列(分别地seqidno:10和11)、pgdm1400scfv核苷酸和aa序列(分别地seqidno:12和13)、pgt128kir核苷酸和aa序列(分别地seqidno:14和15)、pgt128scfv核苷酸和aa序列(分别地seqidno:16和17)、vrc01kir核苷酸和aa序列(分别地seqidno:18和19)、vrc01scfv核苷酸和aa序列(分别地seqidno:20和21)、3bnc60-kir核苷酸和aa序列(分别地seqidno:22和23)、3bnc60scfv核苷酸和aa序列(分别地seqidno:24和25)、vrc01c-mut-kir核苷酸和aa序列(分别地seqidno:26和27)、vrc01c-mutscfv核苷酸和aa序列(分别地seqidno:28和29)、cd4dap12-kirs2核苷酸和aa序列(分别地seqidno:30和31)、cd4-kir核苷酸和aa序列(分别地seqidno:32和33)、和vrc01igg4bbz核苷酸和aa序列(分别地seqidno:34和35)。
图9a-9c是一系列图和直方图,其展现了基于scfv的car(scfv-car)的细胞毒性是特异性的和强力的。vrc01car针对表达hivyu-2毒株的靶标细胞(图9a)而非表达cd19的细胞(图9b)显示了特异性细胞毒性;通过使用杀伤细胞免疫球蛋白样受体(kir)细胞质结构域(图9a)可以增加细胞毒性。杀伤功效与通过cart细胞的干扰素γ(ifng)释放相关联(图9c)。51cr释放试验的持续时间是4小时。kir构建体由dap12和连接至kirs2跨膜结构域的scfv构成。g4-bbz构建体由连接至igg4铰链、cd8跨膜结构域和4-1bb的scfv以及cd3ζ细胞内尾的构成。bn01=vrc01;ntd=未转导的对照t细胞。
图10a-10c是一系列图和直方图,其描绘了scfv-car的细胞特异性细胞毒性的另一个例子。3bnc60和vrc01c-car显示了表达hiv-1包膜gp120/41的k562细胞(yu-2分离株)的特异性裂解(图10a),而非表达cd19的k562细胞(图10b)。如图9c中先前显示的,通过cart细胞的特异性ifnγ产生与杀伤功效相关联(图10c)。3bnc60涉及另一个cd4结合位点广泛中和抗体。vrc01-c是vrc01的衍生物,其在hcdr3和hcdr1之间缺少二硫键并且可以因此与原始的vrc01scfv——其包含另外的、非典型的二硫键——比较显示更大百分比的正确折叠的scfv。对于3bnc60和vrc01-c,在kir和igg4-bbz(也称为g4)构建体之间,在杀伤方面不存在可检测的差异。
图11a-11c是展现cd4car特异性地应答于env+细胞而非ii类mhc+细胞的系列图。图11a:原代人cd8t细胞使用αcd3/αcd28涂覆的珠被活化并且保持未转导的(ntd)或转导ef1α-cd8αtm慢病毒载体——其编码单独表达cd3-ζ信号传导结构域,或与cd28或4-1bb共刺激结构域组合表达cd3-ζ信号传导结构域的cd4car。活化之后两周,cd8t细胞在1:1比率下与未修饰的k562细胞、表达高水平的hla-dr的k562细胞(参见图14)、或表达hiv-1yu2gp160的k562细胞共培养6小时。在左侧上显示细胞内ifnγ和mip-1β表达,和在右侧上显示细胞内il-2表达和cd107a表面动员。图11b:设计共培养试验以展现cd4car+cd8t细胞不杀伤表达ii类mhc的靶标细胞。简略地,来自图6a的ntd或转导cd428zcar的cd8t细胞与表达hla-a2和gfp的k562细胞以及表达hla-dr*0401和mcherry的k562在1:1:1比率下共培养。在混合(0hr)和在3天的共培养(72hr)后立刻进行测量gfp和mcherry表达的流式细胞术。图11c:以一式三份进行的单个实验的汇总数据,其测量表达hla-a2/gfp的细胞与表达hla-dr*0401/mcherry的细胞在24、48、和72小时的培养后的比率。误差棒指示平均值的标准误差(sem)。数据代表三个独立实验。
图12a-12f是描绘表达优化的cd4car的t细胞控制hiv-1复制并且体内扩展至比原始cd4car大得多的水平的系列图。使用8百万个cd4t细胞和2百万个cd8t细胞输注7只nsg(nod-scidil2rgnull)小鼠的组群。cd8t细胞保持未转导的(ntd),转导优化的(ef1α-cd8αtm,慢病毒载体)cd4-ζcar——其包含4-1bb或cd28细胞内共刺激结构域,或转导临床试验(基于mmlv的,pgk-cd4tm)cd4car,其分别表示为ntd、bbz、28z、和cd4z。cd8t细胞转导效率在注入小鼠之前被归一化至50%。在注射之后三周,测量移植物以测定(图12a)基线外周cd4t细胞计数和(图12c)car+cd8t细胞计数。两天之后,经由尾静脉注射以hiv-1bal感染小鼠。感染之后22天,(图12b)获得终点外周cd4t细胞计数和(图12d)car+cd8t细胞计数。感染之后(图12e)七天和(图12f)十八天,测量血浆hivrna的拷贝。秩和检验被用于测定统计学显著性(p值:ns>0.05,*<0.05,**<0.01,***<0.0001)。
图13a-13c是显示体内富集ccr5-zfn修饰的cd4carcd8t细胞的系列图。在图7中描述的nsg小鼠实验设计中,添加四个另外的组群,其中cd8t细胞在活化和cd4car转导前48小时电穿孔有ccr5zfnrna,或保持未转导的(ntd)。图13a:在22天的hiv感染后,脾cd8t细胞从感染hiv的小鼠被分离并且分析ccr5破坏频率。图13b:通过人cd4和cd8染色和对在hiv或模拟对照感染之后22天分离的外周血进行流式细胞术来测定表达cd4car的cd8t细胞与未转导的cd8t细胞的比率。流式细胞术图显示在图15中。图13c:感染之后十八天,在感染hiv的小鼠中测量血浆hivrna的拷贝,如图12f中,但是具有并入的四个zfn处理组。使用秩和检验检测显著性(p值:ns>0.05,*<0.05,**<0.01,***<0.0001)。
图14是描绘转导hla-dr的k562细胞上高水平的ii类mhc表达的图。为了确认转导hla-dr*0401的k562细胞上ii类mhc的高表达,通过流式细胞术测量hla-dr表达。作为门控对照,已经转导hla-a2的k562细胞被染色,以及表达高的ii类mhc的rajib细胞。描绘了覆盖三种细胞群的直方图。
图15是一系列图,其描绘了4-1bb共刺激结构域在不存在抗原的情况下导致较大的t细胞持续性。在22天的模拟对照感染后,来自转导bbz或28z的、zfn处理的小鼠组群的外周血细胞或脾细胞使用αcd4和αcd8抗体进行car表达染色。顶图组显示了外周血和底图组显示了脾细胞。在来自感染hiv的小鼠的所有情况中显示了一个代表性流式图(flowplot)。
图16a-16b是展现ccr5zfn处理不改进cd4carcd8t细胞体内保护cd4t细胞或扩展car+cd8t细胞计数的能力的系列图。图16a:在hiv感染之后22天收集终点外周血cd4t细胞计数,如图12b中显示的,但是现在还包括来自接受ccr5zfn修饰的cd8t细胞的小鼠的数据。(b)在所有组群中,在感染之后22天枚举终点外周血car+cd8t细胞计数。使用秩和检验检测显著性(p值:ns>0.05,*<0.05,**<0.01,***<0.0001)。
图17a-17d是概述本发明中对原始临床试验载体做出的改进的系列表格和图。图17a:描绘探索以改进原始临床试验mmlv基构建体的修饰的完整列表的表格和示意图。图17b:使用在图2c中概述的实验设计,原代人cd8t细胞使用αcd3/αcd28涂覆的珠被活化并且保持为未转导的(ntd),转导由pgk启动子驱动的基于原始mmlv的cd4基car(临床试验car),或转导放置在hiv基慢病毒载体中的优化的ef1α-cd8αtmcar。转导效率在共培养之前被归一化至60%。在7天的与感染hivbal的cd4t细胞的共培养后,通过流式细胞术——在cd8阴性t细胞上门控——测量表面cd4和细胞内gagp24的表达。图17c:显示在cd8阳性细胞上门控。图17d:在cd8阴性细胞上门控的以一式三份进行的单个实验的汇总数据。误差棒指示平均值的标准误差(sem)。此数据代表3个独立实验。
图18是本发明的优化的hivcd4car和基于hiv抗体的car中的一些的注释的氨基酸序列(seqidno:44-61)的列表。
图19是本发明的优化的hivcd4car和基于hiv抗体的car中的一些的注释的核酸序列(seqidno:63-79)的列表。
具体实施方式
定义
除非另外定义,本文使用的所有技术和科学术语具有与本发明所属领域普通技术人员通常理解的相同含义。虽然与本文描述的那些类似或等价的任何方法和材料可以在实践中被用于测试本发明,但是本文描述了优选的材料和方法。在描述和要求保护本发明中,将使用下列术语。
还理解本文使用的术语仅出于描述特定实施方式的目的,并且不意图是限制性的。
本文使用冠词“一个”和“一种”,指的是一个或多于一个(即,至少一个)该冠词的语法对象。举例而言,“一个要素”意思是一个要素或多于一个要素。
如本文使用的“大约”,当指的是可测量的值比如量、时间期间等时,意思是涵盖从规定值的±20%或±10%、更优选地±5%、甚至更优选地±1%、和还更优选地±0.1%的变化,只要这样的变化适于进行公开的方法。
如本文使用的“活化”指的是已经被充分地刺激以诱导可检测的细胞增殖的t细胞的状态。活化也可以与诱导的细胞因子产生,和可检测的效应物功能相关联。术语“活化的t细胞”指的是经历细胞分裂的t细胞,等等。
如本文使用的,术语“衔接分子”指的是具有如下序列的多肽:其允许与两个或更多个分子相互作用,并且在某些实施方式中,促进细胞毒性细胞的活化或失活。
如本文使用的术语“抗体”指的是与抗原特异性地结合的免疫球蛋白分子。抗体可以是从自然来源或从重组来源获得的完整免疫球蛋白,并且可以是完整免疫球蛋白的免疫反应性部分。抗体通常是免疫球蛋白分子的四聚体。本发明中的抗体可以以多种形式存在,包括,例如,多克隆抗体、单克隆抗体、fv、fab和f(ab)2,以及单链抗体(scfv)和人源化抗体(harlow等1999,in:usingantibodies:alaboratorymanual,coldspringharborlaboratorypress,ny;harlow等1989,in:antibodies:alaboratorymanual,coldspringharbor,newyork;houston等1988,proc.natl.acad.sci.usa85:5879-5883;bird等1988,science242:423-426)。
术语“抗体片段”指的是部分完整抗体,并且指的是完整抗体的抗原决定可变区。抗体片段的实例包括但不限于fab、fab'、f(ab')2、和fv片段,线性抗体,scfv抗体,和由抗体片段形成的多特异性抗体。
如本文使用的“抗体重链”指的是以它们自然存在的构象存在于所有抗体分子中的两种类型的多肽链中较大的链。
如本文使用的“抗体轻链”指的是以它们自然存在的构象存在于所有抗体分子中的两种类型的多肽链中较小的链。α和β轻链指的是两种主要的抗体轻链同种型。
如本文使用的术语“合成抗体”的意思是使用重组dna技术生成的抗体,比如,例如,如本文描述的由噬菌体表达的抗体。术语也应当解释为意思是已经由dna分子——其编码抗体并且该dna分子表达抗体蛋白——或规定抗体的氨基酸序列的合成生成的抗体,其中dna或氨基酸序列已经使用本领域可获得和熟知的合成dna或氨基酸序列技术获得。
如本文使用的术语“抗原”或“ag”被定义为激发免疫应答的分子。此免疫应答可以涉及抗体产生,或特异性免疫活性细胞的活化,或二者。技术人员将理解任何大分子——实际上包括所有蛋白质或肽——可以充当抗原。另外,抗原可以源自重组或基因组dna。技术人员将理解任何dna——其包括编码引起免疫应答的蛋白质的核苷酸序列或部分核苷酸序列——因此编码如本文使用的该术语“抗原”。另外,本领域技术人员将理解抗原不必单独地由基因的全长核苷酸序列编码。容易显而易见的是本发明包括但不限于多于一个基因的部分核苷酸序列的用途,并且这些核苷酸序列以不同的组合布置以引起期望的免疫应答。而且,技术人员将理解抗原完全不必由“基因”编码。容易显而易见的是抗原可以被生成、合成或可以源自生物学样品。这样的生物学样品可以包括但不限于组织样品、肿瘤样品、细胞或生物学流体。
如本文使用的术语“抗肿瘤效应”指的是生物学效应,其可以由肿瘤体积的减小、肿瘤细胞数目的减少、转移数目的减少、预期寿命的增加、或与癌性病症相关联的多种生理学症状的改善显现。“抗肿瘤效应”也可以由本发明的肽、多核苷酸、细胞和抗体在预防肿瘤在第一位置的发生的能力显现。
根据本发明,术语“自身抗原”意思是由免疫系统识别为异物(foreign)的任何自体抗原。自身抗原包括但不限于细胞蛋白、磷蛋白、细胞表面蛋白、细胞脂质、核酸、糖蛋白,包括细胞表面受体。
如本文使用的,术语“自身免疫疾病”被定义为由自身免疫反应导致的障碍。自身免疫疾病是对自体抗原的不适当和过度应答的结果。自身免疫疾病的实例包括但不限于阿狄森氏病、斑秃、强直性脊柱炎、自身免疫性肝炎、自身免疫性腮腺炎、克罗恩病、糖尿病(i型)、附睾炎、肾小球性肾炎、格雷夫斯氏病、格-巴二氏综合征、桥本氏病、溶血性贫血、系统性红斑狼疮、多发性硬化、重症肌无力、寻常天疱疮、牛皮癣、风湿热、类风湿性关节炎、结节病、硬皮病、斯耶格伦氏综合征、脊椎关节病、甲状腺炎、血管炎、白斑病、粘液性水肿、恶性贫血、溃疡性结肠炎等。
如本文使用的,术语“自体的”意思是指源自相同个体的任何物质,其随后被再引入该个体。
“同种异体的”指的是源自相同物种的不同动物的移植物。
“异种的”指的是源自不同物种的动物的移植物。
术语“广泛中和抗体(bnab)”指的是通过中和其效应防护细胞免于特定病毒的多种毒株的抗体。在一些实施方式中,广泛中和hiv-1抗体(bnab)是中和多种hiv-1病毒株的中和抗体。
如本文使用的术语“癌症”被定义为以畸变细胞的快速和失控生长为特征的疾病。癌细胞可以局部地扩散或通过血流和淋巴系统扩散至身体的其它部分。多种癌症的实例包括但不限于乳腺癌、前列腺癌、卵巢癌、宫颈癌、皮肤癌、胰腺癌、结肠直肠癌、肾癌、肝癌、脑癌、淋巴瘤、白血病、肺癌等。
如本文使用的术语“cd4”意思是规定来自任何来源的cd4的任何氨基酸序列,包括已经通过密码子优化编码cd4的核酸序列生成的cd4的氨基酸序列。可以使用设计以优化氨基酸序列中的密码子的任何可用的技术和算法完成密码子优化。
如本文使用的术语“嵌合抗原受体”或“car”指的是被工程化以在免疫效应细胞上表达和特异性地结合抗原的人工t细胞受体。car可以被用作使用过继性细胞转移的疗法。t细胞从患者去除并且被修饰,使得它们表达特异于特定形式的抗原的受体。例如,在一些实施方式中,已经以对肿瘤相关抗原的特异性表达car。car还可以包括细胞内活化结构域、跨膜结构域和细胞外结构域,该细胞外结构域包括肿瘤相关抗原结合区。在一些方面,car包括融合单链可变片段(scfv)衍生的单克隆抗体,其被融合至跨膜和细胞内结构域。car设计的特异性可以源自受体的配体(例如,肽)。在一些实施方式中,通过重定向表达特异于hiv相关抗原的car的t细胞的特异性,car可以靶向感染hiv的细胞。
术语“嵌合细胞内信号传导分子”指的是重组受体,其包括一个或多个共刺激分子的一个或多个细胞内结构域。嵌合细胞内信号传导分子基本上缺少细胞外结构域。在一些实施方式中,嵌合细胞内信号传导分子包括额外的结构域,比如跨膜结构域、可检测的标签、和间隔结构域。
如本文使用的,术语“保守序列修饰”意图指的是氨基酸修饰,其不显著地影响或改变包含该氨基酸序列的抗体的结合特性。这样的保守修饰包括氨基酸置换、添加和缺失。修饰可以通过本领域已知的标准技术引入本发明的抗体,比如定点诱变和pcr介导的诱变。保守氨基酸置换是其中氨基酸残基被替换为具有类似侧链的氨基酸残基的置换。本领域已经定义了具有类似侧链的氨基酸残基的家族。这些家族包括具有碱性侧链(例如,赖氨酸、精氨酸、组氨酸)、酸性侧链(例如,天冬氨酸、谷氨酸)、不带电的极性侧链(例如,甘氨酸、天冬酰胺、谷氨酰胺、丝氨酸、苏氨酸、酪氨酸、半胱氨酸、色氨酸)、非极性侧链(例如,丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸)、β-支化侧链(例如,苏氨酸、缬氨酸、异亮氨酸)和芳香族侧链(例如,酪氨酸、苯丙氨酸、色氨酸、组氨酸)的氨基酸。因而,抗体的cdr区内的一种或多种氨基酸残基可以被替换为来自相同侧链家族的其它氨基酸残基,并且可以使用本文描述的功能性试验测试改变的抗体结合抗原的能力。
如本文使用的术语“共刺激配体”包括特异性地结合t细胞上的关联共刺激分子的抗原呈递细胞(例如,aapc、树突细胞、b细胞等)上的分子,从而除通过例如将tcr/cd3复合体与使用肽负载的mhc分子结合提供的初级信号之外,还提供了介导t细胞应答的信号,该t细胞应答包括但不限于增殖、活化、分化等。共刺激配体可以包括但不限于cd7、b7-1(cd80)、b7-2(cd86)、pd-l1、pd-l2、4-1bbl、ox40l、可诱导的共刺激配体(icos-l)、细胞间粘附分子(icam)、cd30l、cd40、cd70、cd83、hla-g、mica、micb、hvem、淋巴毒素β受体、3/tr6、ilt3、ilt4、hvem、结合toll配体受体的激动剂或抗体和与b7-h3特异性地结合的配体。共刺激配体也涵盖,特别是与存在于t细胞上的共刺激分子特异性地结合的抗体,比如,但不限于cd27、cd28、4-1bb、ox40、cd30、cd40、pd-1、icos、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、和与cd83特异性地结合的配体。
“共刺激分子”指的是与共刺激配体特异性地结合的t细胞上的关联结合配偶体,从而介导通过t细胞的共刺激应答,比如,但不限于增殖。共刺激分子包括但不限于tcr,cd3ζ,cd3γ,cd3δ,cd3ε,cd86,普通的fcrγ,fcrβ(fcεr1b),cd79a,cd79b,fcγriia,dap10,dap12,t细胞受体(tcr),cd27,cd28,4-1bb(cd137),ox40,cd30,cd40,pd-1,icos,淋巴细胞功能相关抗原-1(lfa-1),cd2,cd7,light,nkg2c,b7-h3,与cd83特异性地结合的配体,cds,icam-1,gitr,baffr,hvem(lightr),slamf7,nkp80(klrf1),cd127,cd160,cd19,cd4,cd8α,cd8β,il2rβ,il2rγ,il7rα,itga4,vla1,cd49a,itga4,ia4,cd49d,itga6,vla-6,cd49f,itgad,cd11d,itgae,cd103,itgal,cd11a,lfa-1,itgam,cd11b,itgax,cd11c,itgb1,cd29,itgb2,cd18,lfa-1,itgb7,tnfr2,trance/rankl,dnam1(cd226),slamf4(cd244、2b4),cd84,cd96(tactile),ceacam1,crtam,ly9(cd229),cd160(by55),psgl1,cd100(sema4d),cd69,slamf6(ntb-a、ly108),slam(slamf1、cd150、ipo-3),blame(slamf8),selplg(cd162),ltbr,lat,gads,slp-76,pag/cbp,nkp44,nkp30,nkp46,nkg2d,本文描述的其它共刺激分子,其任何衍生物、变体、或片段,具有相同功能能力的共刺激分子的任何合成序列,和其任意组合。
如本文使用的“共刺激信号”指的是与初级信号组合,比如tcr/cd3连接,导致t细胞增殖和/或关键分子的上调或下调的信号。
术语“细胞毒的”或“细胞毒性”指的是杀伤或损害细胞。在一个实施方式中,修饰的细胞的细胞毒性被改进,例如增加的t细胞的细胞溶解活性。
“疾病”是动物的健康状态,其中动物不能维持稳态,并且其中如果不改善疾病,则动物的健康继续恶化。相比之下,动物中的“障碍”是健康状态,其中动物能够维持稳态,但是其中动物的健康状态与它没有处于该障碍时相比不太有利。保持不治疗,障碍不必然引起动物健康状态的进一步降低。
“有效量”或“治疗有效量”在本文可交换地使用,并且指的是对实现具体的生物学结果或提供治疗或预防益处有效的如本文描述的化合物、制剂、材料、或组合物的量。这样的结果可以包括但不限于抗肿瘤活性,如通过本领域适合的任何手段测定的。
“编码”指的是多核苷酸比如基因、cdna、或mrna中的核苷酸的特异性序列充当模板来合成生物学过程中的其它多聚体和大分子的固有性质,该多聚体和大分子具有核苷酸(即,rrna、trna和mrna)的限定序列或氨基酸的限定序列中的任一种和由其产生的生物学性质。因而,如果对应于基因的mrna的转录和翻译在细胞或其它生物学系统中产生蛋白质,则该基因编码蛋白质。其核苷酸序列同一于mrna序列并且通常在序列表中提供的编码链,和用作基因或cdna转录的模板的非编码链二者,可以被称为编码该基因或cdna的蛋白质或其它产物。
如本文使用的“内源的”指的是来自生物体、细胞、组织或系统或在生物体、细胞、组织或系统内产生的任何物质。
如本文使用的,术语“外源的”指的是从生物体、细胞、组织或系统引入或在生物体、细胞、组织或系统外产生的任何物质。
如本文使用的术语“扩展”指的是数目的增加,如t细胞数目的增加。在一个实施方式中,离体扩展的t细胞的数目相对于培养物中原始存在的数目增加。在另一个实施方式中,离体扩展的t细胞的数目相对于培养物中的其它细胞类型的数目增加。如本文使用的术语“离体”指的是已经从活的生物体(例如,人)去除,并且在生物体外(例如,在培养皿、试管、或生物反应器中)繁殖的细胞。
如本文使用的术语“表达”被定义为由它的启动子驱动的特定核苷酸序列的转录和/或翻译。
“表达载体”指的是包括重组多核苷酸的载体,该重组多核苷酸包括可操作地连接至待表达的核苷酸序列的表达控制序列。表达载体包括足够的用于表达的顺式作用元件;用于表达的其它元件可以由宿主细胞供应或在体外表达系统中供应。表达载体包括所有本领域已知的那些,比如并入重组多核苷酸的粘粒、质粒(例如,裸露的或包含在脂质体中)和病毒(例如,慢病毒、逆转录病毒、腺病毒、和腺伴随病毒)。
如本文使用的术语“人免疫缺陷病毒”或“hiv”意思是本领域已知或迄今未知的任何hiv毒株或变体,非限制性地包括hiv-1和hiv-2。
如本文使用的“同源的”指的是两个聚合物分子之间,例如,两个核酸分子——比如,两个dna分子或两个rna分子——之间,或两个多肽分子之间的亚单元序列同一性。当两个分子二者中的亚单元位置被相同的单体亚单元占据时;例如,如果两个dna分子的每个中的位置被腺嘌呤占据,则它们在该位置处是同源的。两个序列之间的同源性是匹配或同源位置的数目的直接函数;例如,如果两个序列中的一半位置(例如,长度为十个亚单位的聚合物中的五个位置)是同源的,则两个序列是50%同源的;如果90%的位置(例如,10个中的9个)是匹配的或同源的,则两个序列是90%同源的。如适用于核酸或蛋白质,如本文使用的“同源的”指的是具有大约50%序列同一性的序列。更优选地,同源序列具有大约75%序列同一性,甚至更优选地,具有至少大约80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%序列同一性。
非人(例如,鼠)抗体的“人源化”形式的是嵌合免疫球蛋白、免疫球蛋白链或其片段(比如fv、fab、fab'、f(ab')2或抗体的其它抗原-结合子序列),其包含源自非人免疫球蛋白的最小序列。就大部分而言,人源化抗体是人免疫球蛋白(受体抗体),其中来自受体的互补决定区(cdr)的残基被替换为来自非人物种(供体抗体)比如小鼠、大鼠或兔的cdr的残基,其具有期望的特异性、亲和力和能力。在一些情况下,人免疫球蛋白的fv框架区(fr)残基被替换为对应的非人残基。另外,人源化抗体可以包括既没有在受体抗体中也没有在输入的cdr或框架序列中发现的残基。做出这些修饰以进一步改进和优化抗体性能。一般而言,人源化抗体将包括基本上所有至少一种和通常两种可变结构域,其中所有或基本上所有cdr区对应于非人免疫球蛋白的那些并且所有或基本上所有fr区是人免疫球蛋白序列的那些。人源化抗体最佳地还将包括至少部分免疫球蛋白恒定区(fc),通常是人免疫球蛋白的。进一步的细节参见jones等nature,321:522-525,1986;reichmann等nature,332:323-329,1988;presta,curr.op.struct.biol.,2:593-596,1992。
“全人(fullyhuman)”指的是免疫球蛋白,比如抗体,其中完整分子具有人起源或由同一于人形式的抗体的氨基酸序列构成。
如本文使用的“同一性”指的是两个聚合物分子之间,特别是两个氨基酸分子之间,比如,两个多肽分子之间的亚基序列同一性。当两个氨基酸序列在相同的位置处具有相同的残基时;例如,如果两个多肽分子中的每个中的位置均被精氨酸占据,则它们在该位置处是同一的。在比对中,两个氨基酸序列在相同的位置处具有相同残基的同一性或程度经常表达为百分数。两个氨基酸序列之间的同一性是匹配或同一位置的数目的直接函数;例如,如果两个序列中的一半位置(例如,十个氨基酸长度的聚合物中的五个位置)是同一的,则两个序列是50%同一的;如果90%的位置(例如,10个中的9个)是匹配的或同一的,则两个氨基酸序列是90%同一的。
“基本上同一的”的意思是展现与参考氨基酸序列(例如,本文描述的氨基酸序列中的任一种)或核酸序列(例如,本文描述的核酸序列中的任一种)至少50%同一性的多肽或核酸分子。优选地,在氨基酸水平或核酸下,这样的序列至少60%,更优选地80%或85%,和更优选地90%、95%、或甚至99%同一于用于比较的序列。
引导核酸序列可以与双链dna靶标位点的一条链(核苷酸序列)互补。引导核酸序列和靶标序列之间的互补百分数可以是至少50%、51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、61%、62%、63%、63%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%。引导核酸序列的长度可以是至少10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35或更多个核苷酸。在一些实施方式中,引导核酸序列包括10至40个核苷酸的连续区段。可变靶向结构域(variabletargetingdomain)可以由dna序列、rna序列、修饰的dna序列、修饰的rna序列(参见例如本文描述的修饰)、或其任意组合组成。
通常使用序列分析软件(例如,1710universityavenue,madison,wis.53705威斯康星大学生物技术中心遗传学计算机组的序列分析软件程序包、blast、bestfit、gap、或pileup/prettybox程序)测量序列同一性。这样的软件通过向多种置换、缺失、和/或其它修饰分配同源性程度来匹配同一的或相似的序列。保守性置换通常包括在下列组内的置换:甘氨酸,丙氨酸;缬氨酸,异亮氨酸,亮氨酸;天冬氨酸,谷氨酸,天冬酰胺,谷氨酰胺;丝氨酸,苏氨酸;赖氨酸,精氨酸;和苯丙氨酸,酪氨酸。在测定同一性程度的示例性方法中,可以使用blast程序,其中e-3和e-100之间的可能性分数指示密切相关的序列。
如本文使用的术语“免疫球蛋白”或“ig”被定义为起抗体作用的一类蛋白质。由b细胞表达的抗体有时被称为bcr(b细胞受体)或抗原受体。包括在此类蛋白质中的五个成员是iga、igg、igm、igd、和ige。iga是存在于身体分泌物比如唾液、泪液、母乳、胃肠分泌物和呼吸道与泌尿生殖道的粘液分泌物中的初次抗体。igg是最常见的循环抗体。igm是在大多数对象中的初次免疫应答中产生的主要免疫球蛋白。它在凝集、补体结合、和其它抗体应答中是最有效的免疫球蛋白,并且在抵御细菌和病毒方面是重要的。igd是不具有已知抗体功能的免疫球蛋白,但是可以充当抗原受体。ige是在暴露于过敏原之后,通过引起从肥大细胞和嗜碱性粒细胞释放介体,介导速发过敏性的免疫球蛋白。
如本文使用的术语“免疫应答”被定义为当淋巴细胞将抗原分子识别为异物和诱发形成抗体和/或活化淋巴细胞以去除抗原时发生的对抗原的细胞应答。
如本文使用的,“指导材料”包括出版物、记录、图表、或任何其它可以用于传达本发明的组合物和方法的有用性的表达媒介。本发明的试剂盒的指导材料可以例如被附加至包含本发明的核酸、肽、和/或组合物的容器,或与包含核酸、肽、和/或组合物的容器一起运送。可选地,指导材料可以与容器分开地运送,目的是指导材料和化合物由接受者配合使用。
“分离的”意思是从自然状态改变或去除。例如,天然存在于活动物中的核酸或肽不是“分离的”,但是部分或完全与它的自然状态的共存物质分离的相同的核酸或肽是“分离的”。分离的核酸或蛋白质可以以基本上纯化的形式存在,或可以存在于非自然环境,比如,例如,宿主细胞。
“kir”意思是杀伤细胞免疫球蛋白样受体,kir已经在人和非人灵长类中被表征,并且是在淋巴细胞——包括nk细胞和一些t细胞——的某些亚单位上存在的多态性1型跨膜分子。kir通过与i类mhc分子的α1和2结构域中的决定簇相互作用调控nk细胞的杀伤功能。此相互作用允许它们检测感染病毒的细胞或肿瘤细胞。大多数kir是抑制性的,意思是它们的mhc识别阻抑表达它们的nk细胞的细胞毒活性。仅有限数目的kir具有活化细胞的能力。kir基因家族具有在位于19号染色体上的白细胞受体复合体(lrc)(19q13.4)的100-200kb区内编码的至少15个基因座(kir2dl1、kir2dl2/l3、kir2dl4、kir2dl5a、kir2dl5b、kir2ds1、kir2ds2、kir2ds3、kir2ds4、kir2ds5、kir3dl1/s1、kir3dl2、kir3dl3)和两个假基因(kir2dp1和kir3dp1)。lrc构成快速进化免疫基因的大的(1mb)和致密的簇,其包含编码具有不同的ig样细胞外结构域的其它细胞表面分子的基因。此外,延长的lrc包含编码跨膜衔接分子dap10和dap12的基因。
如本文使用的“慢病毒”指的是逆转录病毒科家族的属。在逆转录病毒中慢病毒是唯一能够感染非分裂细胞的;它们可以递送显著量的遗传信息进入宿主细胞的dna,以便它们是基因递送载体的最有效的方法之一。hiv、siv、和fiv是慢病毒的所有实例。源自慢病毒的载体提供了实现显著水平的基因体内转移的工具。
如本文使用的术语“修饰的”意思是本发明的分子或细胞的改变的状态或结构。分子可以以许多方式被修饰,包括化学地、结构地和功能地。细胞可以通过引入核酸被修饰。
如本文使用的术语“调节”意思是与不存在治疗或化合物的对象中的应答水平比较,和/或与以其它方式相同但未治疗的对象中的应答水平比较,介导对象中的应答水平中的可检测的增加或减少。术语涵盖扰乱和/或影响天然信号或应答,从而介导对象(优选地,人)中的有益的治疗性应答。
在本发明的背景下,使用常见核酸碱基的下列缩写。“a”指的是腺苷,“c”指的是胞嘧啶,“g”指的是鸟苷,“t”指的是胸苷,和“u”指的是尿苷。
除非另外规定,“编码氨基酸序列的核苷酸序列”包括是彼此的简并译本并且编码相同的氨基酸序列的所有核苷酸序列。短语编码蛋白质或rna的核苷酸序列还可以包括内含子,其程度为编码该蛋白质的核苷酸序列可以在一些译本中包含内含子(一个或多个)。
术语“可操作地连接”指的是调控序列和异源核酸序列之间的功能连接,其导致后者的表达。例如,当第一核酸序列处于与第二核酸序列的功能关系中时,第一核酸序列与第二核酸序列可操作地连接。例如,如果启动子影响编码序列的转录或表达,则启动子被可操作地连接至编码序列。通常,可操作地连接的dna序列是连续的,并且其中在相同的阅读框中必须接合两个蛋白质编码区。
术语“过表达的”肿瘤抗原或肿瘤抗原的“过表达”意图指示相对于来自组织或器官的正常细胞的表达水平,来自疾病区域如患者的特定组织或器官内的实体瘤的细胞中的肿瘤抗原的的异常表达水平。患有以肿瘤抗原的过表达为特征的实体瘤或血液学恶性肿瘤的患者可以由本领域已知的标准试验确定。
免疫原性组合物的“肠胃外”施用包括,例如,皮下(s.c.)、静脉内(i.v.)、肌肉内(i.m.)、或胸骨内注射,或输注技术。
如本文使用的术语“多核苷酸”被定义为核苷酸链。另外,核酸是核苷酸的聚合物。因而,如本文使用的核酸和多核苷酸是可交换的。本领域技术人员具有核酸是可以被水解为单体“核苷酸”的多核苷酸的一般知识。单体核苷酸可以被水解为核苷。如本文使用的多核苷酸包括但不限于通过本领域可获得的任何手段——非限制性地包括重组手段,即,使用普通克隆技术和pcrtm等从重组文库或细胞基因组克隆核酸序列——和通过合成手段获得的所有核酸序列。
如本文使用的,术语“肽”、“多肽”和“蛋白质”可交换地使用,并且指的是由肽键共价连接的氨基酸残基组成的化合物。蛋白质或肽必须包含至少两个氨基酸,并且对可以包括蛋白质或肽的序列的氨基酸的最大数目没有限制。多肽包括任何肽或蛋白质,该肽或蛋白质包括通过肽键相互接合的两个或更多个氨基酸。如本文使用的,术语指的是短链,其在本领域中也通常被称为例如肽、寡肽和寡聚物;和较长链二者,其在本领域中通常被称为蛋白质,其具有许多类型。“多肽”包括例如生物学活性片段、基本上同源的多肽、寡肽、同二聚体、异二聚体、多肽的变体、修饰的多肽、衍生物、类似物、融合蛋白等。多肽包括天然肽、重组肽、合成肽、或其组合。
如本文使用的术语“启动子”被定义为起始多核苷酸序列的特异性转录需要的,由细胞的合成机器识别,或引导合成机器的dna序列。
如本文使用的,术语“启动子/调控序列”意思是可操作地连接至启动子/调控序列的基因产物的表达需要的核酸序列。在一些情况下,此序列可以是核心启动子序列,并且在其它情况下,此序列还可以包括基因产物的表达需要的增强子序列和其它调控元件。例如,启动子/调控序列可以是以组织特异性方式表达基因产物的序列。
“组成型”启动子是如下核苷酸序列,当其与编码或规定基因产物的多核苷酸可操作地连接时,引起在细胞的大多数或所有生理学条件下在细胞中产生基因产物。
“诱导型”启动子是如下核苷酸序列,当其与编码或规定基因产物的多核苷酸可操作地连接时,引起在基本上仅当对应于启动子的诱导物存在于细胞中时,在细胞中产生基因产物。
“组织-特异性”启动子是如下核苷酸序列,当其与编码基因或由基因规定的多核苷酸可操作地连接时,引起基本上只要细胞是对应于相应于启动子的组织类型的细胞,则在细胞中产生基因产物。
术语“对免疫抑制的抗性”指的是缺少阻抑或降低的免疫系统活性或活化的阻抑。
“信号转导途径”指的是各种信号转导分子——其在传递来自细胞的一部分的信号至细胞的另一部分中发挥作用——之间的生物化学关系。短语“细胞表面受体”包括分子和分子的复合体,其能够接收信号和跨越细胞的质膜传递信号。
“单链抗体”指的是通过重组dna技术形成的抗体,其中免疫球蛋白重链和轻链片段经由工程化跨度的氨基酸连接至fv区。生成单链抗体的多种方法是已知的,包括在美国专利号4,694,778;bird(1988)science242:423-442;huston等(1988)proc.natl.acad.sci.usa85:5879-5883;ward等(1989)nature334:54454;skerra等(1988)science242:1038-1041中描述的那些。
如本文使用的关于抗体的术语“特异性地结合”意思是识别特异性抗原但是基本上不识别或结合样品中的其它分子的抗体。例如,特异性地结合至来自一个物种的抗原的抗体也可以结合至来自一个或多个物种的该抗原。但是,这样的跨物种反应性本身不将抗体的类别改变为特异性的。在另一个实例中,特异性地结合至抗原的抗体也可以结合至抗原的不同等位基因形式。然而,这样的交叉反应性本身不将抗体的类别改变为特异性的。在一些情况下,术语“特异性结合”或“特异性地结合”可以关于抗体、蛋白质、或肽与第二化学种类的相互作用使用,意思是相互作用依赖化学种类上特定结构(例如,抗原决定簇或表位)的存在;例如,抗体识别和结合至特异性蛋白质结构,而不是一般地识别和结合至蛋白质。如果抗体特异于表位“a”,则在包含标示的“a”和抗体的反应中存在包含表位a(或游离的、未标示的a)的分子,将降低结合至抗体的标示的a的量。
术语“刺激”意思是通过结合刺激分子(例如,tcr/cd3复合体)与其关联配体,从而介导信号转导事件——比如,但不限于经由tcr/cd3复合体的信号转导——诱导的初次应答。刺激可以介导某些分子的改变的表达,比如tgf-β的下调、和/或细胞骨架结构的重组等。
“刺激分子”,作为本文使用的术语,意思是与存在于抗原呈递细胞上的关联刺激配体特异性地结合的t细胞上的分子。
如本文使用的“刺激配体”意思是如下配体:其当存在于抗原呈递细胞(例如,aapc、树突细胞、b细胞等)上时,可以与t细胞上的关联结合配偶体(在本文称为“刺激分子”)特异性地结合,从而介导t细胞的初次应答,包括但不限于活化、免疫应答的起始、增殖等。刺激配体在本领域是熟知的,并且涵盖,特别是使用肽、抗cd3抗体、超激动剂抗cd28抗体、和超激动剂抗cd2抗体负载的i类mhc分子。
术语“对象”意图包括其中可以引发免疫应答的活的生物体(例如,哺乳动物)。如其中使用的“对象”或“患者”可以是人或非人哺乳动物。非人哺乳动物包括,例如,家畜和宠物,比如羊、牛、猪、狗、猫和鼠哺乳动物。优选地,对象是人。
如本文使用的,术语“基本上缺少细胞外结构域”指的是基本上不含细胞外突出(extrude)的结构域的分子。在一个实施方式中,嵌合细胞内信号传导分子缺少通过细胞外结构域执行的任何功能,比如抗原结合。在另一个实施方式中,嵌合细胞内信号传导分子包括跨膜结构域但是缺少功能性细胞外结构域。
如本文使用的,“基本上纯化的”细胞是基本上不含其它细胞类型的细胞。基本上纯化的细胞也指的是已经与其它细胞类型——在其天然存在状态中与该其它细胞类型正常地相关联——分离的细胞。在一些情况下,基本上纯化的细胞群指的是均质细胞群。在其它情况下,此术语简单地指的是已经与如下细胞分离的细胞:在其天然状态中与该细胞正常地相关联。在一些实施方式中,在体外培养细胞。在其它实施方式中,不在体外培养细胞。
“靶标位点”或“靶标序列”指的是基因组核酸序列,其限定了在足以发生结合的条件下结合分子可以特异性地结合至其的部分核酸。
如本文使用的,术语“t细胞受体”或“tcr”指的是响应于抗原的呈递参与t细胞的活化的膜蛋白的复合体。tcr负责识别结合至主要组织相容性复合体分子的抗原。tcr由α和β链的异二聚体组成,但是在一些细胞中,tcr由γ和δ(γ/δ)链构成。tcr可以以α/β和γ/δ形式存在,其是结构上相似的,但是具有不同的解剖学位置和功能。每条链由两个细胞外结构域——可变和恒定结构域——组成。在一些实施方式中,tcr可以在包括tcr的任何细胞上被修饰,包括,例如,辅助性t细胞、细胞毒性t细胞、记忆t细胞、调节性t细胞、自然杀伤t细胞、和γδt细胞。
如本文使用的术语“治疗性的”意思是治疗和/或预防。治疗性效果通过疾病状态的阻抑、缓解、或根除获得。
如本文使用的术语“转染的”或“转化的”或“转导的”指的是如下过程:通过该过程外源性核酸被转移或引入宿主细胞。“转染的”或“转化的”或“转导的”细胞是已经被转染、转化或转导外源性核酸的细胞。细胞包括原代对象细胞和其子代。
“治疗”疾病,作为本文使用的术语,意思是降低对象经历的疾病或障碍的至少一种迹象或症状的频率或严重度。
如本文使用的术语“肿瘤”指的是异常生长的组织,其可以是良性的、癌前的、恶性的、或转移的。
如本文使用的短语“在转录控制下”或“可操作地连接的”意思是启动子处于与多核苷酸有关的正确的位置和取向,以控制通过rna聚合酶的转录的起始和多核苷酸的表达。
“载体”是物质组合物,其包括分离的核酸,并且其可以被用于递送分离的核酸至细胞内部。众多载体在本领域是已知的,包括但不限于线性多核苷酸、与离子或两亲性化合物相关联的多核苷酸、质粒、和病毒。因而,术语“载体”包括自主复制的质粒或病毒。术语也应当解释为包括便于将核酸转移入细胞的非质粒和非病毒化合物,比如,例如,聚赖氨酸化合物、脂质体等。病毒载体的实例包括但不限于腺病毒载体、腺伴随病毒载体、逆转录病毒载体、慢病毒载体等。
范围:遍及本公开内容,本发明的多个方面可以以范围格式呈现。应当理解范围格式的描述仅仅出于方便和简洁,并且不应当解释为对本发明的范围的僵硬限制。因此,范围的描述应当被认为已经具体地公开了所有可能的子范围以及该范围内的单个数值。例如,比如1至6的范围的描述应当被认为已经具体地公开了子范围比如1至3、1至4、1至5、2至4、2至6、3至6等,以及该范围内的单个数字,例如,1、2、2.7、3、4、5、5.3和6。不管范围的宽度,这均适用。
描述
本发明包括使用cd4膜结合嵌合受体或hiv特异性scfv嵌合抗原受体(car)治疗hiv感染的组合物和方法。根据本发明,通过表达能够特异性地识别和结合感染hiv的细胞的cd4或scfv的片段,t细胞被修饰用于过继性t细胞疗法。本发明的修饰的t细胞特异于感染hiv的细胞并且针对hiv感染具有改进的细胞毒性和功效。
hiv特异性cd4膜结合嵌合受体
本发明包括膜结合嵌合受体,其包括cd4结构域,具体地特异性地结合至hiv病毒粒子或感染hiv的细胞的cd4细胞外结构域。在某些实施方式中,本发明的cd4膜结合嵌合受体包括特定的结构特征,比如本文公开的特定的氨基酸序列或肽。本发明还包括制造这样的受体的方法。本发明的膜结合嵌合受体可以被并入药物组合物,用于治疗对象,例如用作免疫疗法。因此,本发明提供了用于治疗hiv感染或hiv相关疾病的组合物和方法。
在一方面,本发明包括分离的核酸序列,其编码包括cd4细胞外结构域、跨膜结构域、和信号传导结构域的膜结合嵌合受体,其中cd4细胞外结构域能够识别和结合感染hiv的细胞。在另一方面,本发明包括分离的氨基酸序列,其编码包括cd4细胞外结构域、跨膜结构域、和信号传导结构域的膜结合嵌合受体,其中cd4细胞外结构域能够识别和结合感染hiv的细胞。
在一个实施方式中,cd4细胞外结构域包括seqidno:3、46或64,跨膜结构域包括cd8α铰链(seqidno:4、47或65)和跨膜结构域包括选自cd8α跨膜结构域(seqidno:5、48或66)和cd28跨膜结构域(seqidno:8、49或67)的至少一种结构域。在另一个实施方式中,信号传导结构域包括cd3ζ信号传导结构域(seqidno:6、51或68)。在又另一个实施方式中,共刺激信号传导区包括选自cd27、cd28、4-lbb、ox40、cd30、cd40、pd-1、icos、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、与cd83特异性地结合的配体、和其任意组合的共刺激分子的细胞内结构域。在又另一个实施方式中,共刺激信号传导区cd28(seqidno:9、49或67)存在于构建体中。在进一步的实施方式中,cd4细胞外结构域特异性地结合至hiv包膜(env)糖蛋白。
在一方面,本发明包括载体,其包括真核延伸因子(ef1α)启动子、cd4细胞外结构域、cd8α铰链、cd8α跨膜结构域和cd3ζ信号传导结构域(seqidno:1)。在另一方面,本发明包括载体,其包括ef1α启动子、cd4细胞外结构域、cd8α铰链、cd28跨膜结构域、cd28共刺激结构域和cd3ζ信号传导结构域(seqidno:7)。在又另一个方面,本发明包括载体,其包括膜结合嵌合受体,该膜结合嵌合受体包括cd4细胞外结构域、跨膜结构域、和信号传导结构域,其中cd4细胞外结构域能够识别和结合感染hiv的细胞,其中载体包括选自seqidno:44、45、62和63的至少一种氨基酸序列。
在一个实施方式中,膜结合嵌合受体由seqidno:30、32或64的核酸序列编码。在另一个实施方式中,膜结合嵌合受体具有seqidno:31、33或46的氨基酸序列。
cd4细胞外结构域
cd4是免疫球蛋白超家族的成员并且包括四个细胞外免疫球蛋白结构域(d1至d4)。d1和d3类似于免疫球蛋白可变结构域并且d2和d4类似于免疫球蛋白恒定结构域。d1包括与主要组织相容性复合体ii类分子的β2-微球蛋白相互作用的cd4的区。
在一个实施方式中,膜结合嵌合受体包括cd4的细胞外结构域或其片段。在另一个实施方式中,膜结合嵌合受体包括cd4的至少一个免疫球蛋白结构域。在另一个实施方式中,cd4细胞外结构域包括seqidno:3或64。
本文描述的cd4细胞外结构域,比如cd4的至少一个免疫球蛋白结构域或包括seqidno:3或64的cd4细胞外结构域,可以与本文描述的任何跨膜结构域、本文描述的任何信号传导结构域、或本文描述的可以包括在膜结合嵌合受体中的任何其它结构域组合。
跨膜结构域
在一个实施方式中,跨膜结构域与cd4嵌合抗原受体构建体中的一个结构域缔合。在一些情况下,可以选择跨膜结构域或通过氨基酸置换修饰跨膜结构域,以避免将这样的结构域结合至相同或不同表面膜蛋白的跨膜结构域,从而最小化与受体复合体的其它成员的相互作用。
跨膜结构域可以源自天然来源或合成来源。当来源是天然的时,结构域可以源自任何膜结合蛋白或跨膜蛋白。特定用途的跨膜区可以源自t-细胞受体的α、β或ζ链,cd28,cd3ε,cd45,cd4,cd5,cd8,cd9,cd16,cd22,cd33,cd37,cd64,cd80,cd86,cd134,cd137,cd154(即至少包括上述中的跨膜区(一个或多个))。在一些情况下,也可以采用各种人铰链,包括人ig(免疫球蛋白)铰链。在一个实施方式中,跨膜结构域包括cd8α铰链和跨膜结构域。
在一个实施方式中,跨膜结构域包括cd8α跨膜结构域(seqidno:5或66)。在另一个实施方式中,跨膜结构域包括cd28跨膜结构域(seqidno:8或67)。在又另一个实施方式中,跨膜结构域包括铰链结构域,比如cd8α铰链(seqidno:4或65)。跨膜结构域可以与任何铰链结构域组合和/或可以包括本文描述的一种或多种跨膜结构域。
本文描述的跨膜结构域,比如t-细胞受体的α、β或ζ链,cd28,cd3ε,cd45,cd4,cd5,cd8,cd9,cd16,cd22,cd33,cd37,cd64,cd80,cd86,cd134,cd137,或cd154的至少跨膜区(一种或多种),可以与本文描述的任何cd4细胞外结构域、本文描述的任何信号传导结构域、或可以包括在膜结合嵌合受体中的本文描述的任何其它结构域组合。
在另一个实施方式中,跨膜结构域可以是合成的,在该情况下,它包括占主导的疏水残基比如亮氨酸和缬氨酸。优选地,苯丙氨酸、色氨酸和缬氨酸的三联体存在于合成跨膜结构域的每端处。任选地,短的寡肽或多肽连接体,长度优选地在2和10个氨基酸之间可以在跨膜结构域和细胞质信号传导结构域之间形成连接。甘氨酸-丝氨酸双联体提供了特别合适的连接体。
信号传导结构域
信号传导结构域或细胞内信号传导结构域负责活化其中表达cd4嵌合抗原受体构建体的免疫细胞的正常效应物功能中的至少一种。术语“效应物功能”指的是特定的细胞功能。t细胞的效应物功能,例如,可以是细胞溶解活性或辅助性活性,包括分泌细胞因子。因而术语“细胞内信号传导结构域”或“信号传导结构域”指的是转导效应物功能信号和指导细胞执行特定功能的部分蛋白质。虽然通常可以采用整个信号传导结构域,但是在许多情况下,没有必要使用整个分子。就使用信号传导结构域的截短部分而言,可以替代完整的链使用这样的截短部分,只要它转导效应物功能信号。术语信号传导结构域因而意思是包括足以传导效应物功能信号的信号传导结构域的任何截短部分。
用于本发明的信号传导结构域的实例包括t细胞受体(tcr)和一致作用以在抗原受体衔接之后引发信号转导的共受体的细胞质序列,以及这些序列的任何衍生物或变体和具有相同功能能力的任何合成序列。
已知通过单独的tcr生成的信号不足以用于t细胞的完全活化并且还需要次要或共刺激信号。因而,t细胞活化可以据说由两种不同种类的细胞质信号传导序列介导:通过tcr起始抗原依赖性主要活化的那些(主要细胞质信号传导序列)和以抗原独立性方式作用以提供次要或共刺激信号的那些(次要细胞质信号传导序列)。
主要细胞质信号传导序列以刺激性方式或以抑制性方式调控tcr复合体的主要活化。以刺激性方式作用的主要细胞质信号传导序列可以包含信号传导基序,其称为基于免疫受体酪氨酸的活化基序或itam。
在本发明中具有特别用途的包含主要细胞质信号传导序列的itam的实例包括源自tcrζ、fcrγ、fcrβ、cd3γ、cd3δ、cd3ε、cd5、cd22、cd79a、cd79b、和cd66d的那些。特别优选的是本发明的免疫受体中的细胞质信号传导分子包括源自cd3-ζ的细胞质信号传导序列。
在优选的实施方式中,cd4嵌合抗原受体构建体的信号传导结构域可以被设计以包括单独的cd3-ζ信号传导结构域或连同在免疫受体的背景下有用的任何其它期望的信号传导结构域(一个或多个)。例如,免疫受体的信号传导结构域可以包括cd3ζ链部分和共刺激信号传导区。共刺激信号传导区指的是包括共刺激分子的细胞内结构域的部分免疫受体。共刺激分子是除抗原受体或其配体以外的细胞表面分子,其是淋巴细胞对抗原的高效应答所需的。这样的分子的实例包括cd27、cd28、4-1bb(cd137)、dap12、ox9、ox40、cd30、cd40、pd-1、icos、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、和与cd83特异性地结合的配体等。因而,虽然本发明主要将cd28示例为共刺激信号传导元件,但是其它共刺激元件也在本发明的范围内。
在一个实施方式中,信号传导结构域包括cd3ζ信号传导结构域(seqidno:6、51或68)。在另一个实施方式中,信号传导结构域包括cd4cd28ζ(seqidno:7或62)。在另一个实施方式中,信号传导结构域包括cd28共刺激结构域(seqidno:9或67)。在又另一个实施方式中,信号传导结构域包括cd44-1bb共刺激结构域(seqidno:63)。在又另一个实施方式中,信号传导结构域包括4-1bb(cd137)共刺激结构域(seqidno:69)。信号传导结构域可以包括本文描述的一种或多种信号传导结构域。
本文描述的信号传导结构域,比如tcrζ的α、β或ζ链,fcrγ,fcrβ,cd3γ,cd3δ,cd3ε,cd5,cd22,cd79a,cd79b,或cd66d的细胞质信号传导序列或来自cd27,cd28,4-1bb(cd137),dap12,ox9,ox40,cd30,cd40,pd-1,icos,淋巴细胞功能相关抗原-1(lfa-1),cd2,cd7,light,nkg2c,b7-h3,或与cd83特异性地结合的配体的共刺激信号传导区,可以与本文描述的任何cd4细胞外结构域、本文描述的任何跨膜结构域、或可以包括在膜结合嵌合受体中的本文描述的任何其它结构域组合。
嵌合抗原受体(car)
不希望受限于理论,应理解,使用靶向hiv的car——其中car包括抗-hivscfv——将有益于治疗具有感染hiv的细胞的储存群体(reservoirpopulation)的对象。本发明因此在本发明的一方面包括用于这样的用途的这样的car。
因而,在本发明的一方面,通过在其中表达car来生成t细胞。因而,本发明涵盖car和编码car的核酸构建体,其中car包括抗原结合结构域(例如,编码抗-hiv抗体的scfv)、跨膜结构域和细胞内结构域。
在一方面,本发明包括修饰的细胞,其包括嵌合抗原受体(car),其中car包括抗原结合结构域、跨膜结构域和共刺激分子的细胞内结构域,并且其中细胞是具备靶向的效应物活性的t细胞。在另一方面,本发明包括修饰的细胞,其包括编码嵌合抗原受体(car)的核酸序列,其中核酸序列包括编码抗原结合结构域的核酸序列、编码跨膜结构域的核酸序列和编码共刺激分子的细胞内结构域的核酸序列,并且其中细胞是表达car并且具备靶向的效应物活性(例如靶向的细胞细胞毒性和抗原呈递)的t细胞。在一个实施方式中,靶向的效应物活性针对特异性地结合car的抗原结合结构域的靶标细胞上的抗原。在一方面,靶标抗原是hiv特异性抗原和car包括hiv特异性结合结构域,其包括抗-hiv抗体或其片段。
在一个实施方式中,car由选自seqidno:10、14、18、22、26、34、70-73和74的核酸序列编码。在另一个实施方式中,car具有seqidno:11、15、19、23、27、35、52-55、或56的氨基酸序列。
抗原结合结构域
在一个实施方式中,本发明的抗原结合结构域包括结合至hiv病毒粒子比如hiv包膜糖蛋白(env)(例如gp120)和/或感染hiv的细胞的hiv特异性结合结构域。可以充当抗原的其它细胞表面标志物的实例包括与病毒、细菌和寄生虫感染、自身免疫性疾病、和癌细胞相关联的那些。在另一个实施方式中,本发明的抗原结合结构域包括结合至hiv蛋白的抗体或其片段,比如scfv抗体。优选地,抗原结合结构域是结合至hiv蛋白例如hivenv的scfv抗体。有用的具体scfvs是在图4a、图8、图18和图19中显示的那些(seqidno:11、12、16、17、20、21、24、25、28、29、57-61、75-78、或79)。
抗原结合结构域的选择取决于在靶标细胞的表面上存在的抗原的类型和数目。例如,可以选择抗原结合结构域以识别充当与具体疾病状态相关联的靶标细胞上的细胞表面标志物的抗原。
抗原结合结构域可以包括结合至抗原的任何结构域并且可以包括但不限于单克隆抗体、多克隆抗体、合成抗体、人抗体、人源化抗体、非人抗体、和其任意片段。因而,在一个实施方式中,抗原结合结构域部分包括哺乳动物抗体或其片段。在另一个实施方式中,car的抗原结合结构域选自抗-hiv抗体和其片段。
在一些情况下,抗原结合结构域源自car将最终用于其中的相同的物种是有利的。例如,对于用于人,可能有利的是,car的抗原结合结构域包括如本文其它地方描述的人抗体、或其片段。
还有利的是,抗原结合结构域被可操作地连接至car的另一个结构域,比如跨膜结构域或细胞内结构域,其均在本文其它地方描述,用于在细胞中表达。在一个实施方式中,编码抗原结合结构域的核酸被可操作地连接至编码跨膜结构域的核酸和编码细胞内结构域的核酸。
本文描述的抗原结合结构域,比如结合至hiv蛋白的抗体或其片段,可以与本文描述的任何跨膜结构域、本文描述的任何细胞内结构域或细胞质结构域,或可以包括在car中的本文描述的任何其它结构域组合。
跨膜结构域
关于跨膜结构域,car(或膜结合嵌合受体构建体)可以被设计以包括将car的抗原结合结构域连接至细胞内结构域的跨膜结构域。在一个实施方式中,跨膜结构域天然地与car中的结构域中的一个或多个缔合。在一些情况下,可以选择跨膜结构域或通过氨基酸置换修饰跨膜结构域,以避免将这样的结构域结合至相同或不同表面膜蛋白的跨膜结构域,从而最小化与受体复合体的其它成员的相互作用。
跨膜结构域可以源自天然来源或合成来源。当来源是天然的时,结构域可以源自任何膜结合蛋白或跨膜蛋白。具体用于本发明的跨膜区可以源自t细胞受体的α、β或ζ链,cd28,cd3ε,cd45,cd4,cd5,cd8,cd9,cd16,cd22,cd33,cd37,cd64,cd80,cd86,cd134,cd137,cd154,toll样受体1(tlr1),tlr2,tlr3,tlr4,tlr5,tlr6,tlr7,tlr8,和tlr9(即至少包括上述中的跨膜区(一个或多个))。在一些情况下,也可以采用各种人铰链,包括ig(免疫球蛋白)铰链。
在一个实施方式中,跨膜结构域包括cd8α跨膜结构域(seqidno:5、48、66)。在另一个实施方式中,跨膜结构域包括cd28跨膜结构域(seqidno:8、49和67)。在又另一个实施方式中,跨膜结构域包括铰链结构域,比如cd8α铰链(seqidno:4、47和65)。跨膜结构域可以与任何铰链结构域组合和/或可以包括本文描述的一种或多种跨膜结构域。
本文描述的跨膜结构域,比如t-细胞受体的α、β或ζ链,cd28,cd3ε,cd45,cd4,cd5,cd8,cd9,cd16,cd22,cd33,cd37,cd64,cd80,cd86,cd134,cd137,cd154,toll样受体1(tlr1),tlr2,tlr3,tlr4,tlr5,tlr6,tlr7,tlr8,和tlr9的跨膜区,可以与本文描述的任何抗原结合结构域、本文描述的任何细胞内结构域或细胞质结构域、或可以包括在car中的本文描述的任何其它结构域组合。
在一个实施方式中,跨膜结构域可以是合成的,在该情况下,它将包括占主导的疏水残基比如亮氨酸和缬氨酸。优选地,将在合成跨膜结构域的每端处发现苯丙氨酸、色氨酸和缬氨酸的三联体。
细胞内结构域
car(或膜结合嵌合受体构建体)的细胞内结构域或另外的细胞质结构域包括与在本文其它地方描述的嵌合细胞内信号传导分子类似或相同的细胞内结构域,并且负责活化其中表达car的细胞。
在一个实施方式中,car的细胞内结构域包括负责信号活化和/或转导的结构域。
用于本发明的细胞内结构域的实例包括但不限于表面受体的细胞质部分、共刺激分子、和一致作用以在t细胞中起始信号转导的任何分子,以及这些元件的任何衍生物或变体和具有相同功能能力的任何合成序列。
细胞内结构域的实例包括来自一种或多种分子或受体的片段或结构域,该分子或受体包括但不限于tcr,cd3ζ,cd3γ,cd3δ,cd3ε,cd86,普通的fcrγ,fcrβ(fcεr1b),cd79a,cd79b,fcγriia,dap10,dap12,t细胞受体(tcr),cd8,cd27,cd28,4-1bb(cd137),ox9,ox40,cd30,cd40,pd-1,icos,kir家族蛋白,淋巴细胞功能相关抗原-1(lfa-1),cd2,cd7,light,nkg2c,b7-h3,与cd83特异性地结合的配体,cds,icam-1,gitr,baffr,hvem(lightr),slamf7,nkp80(klrf1),cd127,cd160,cd19,cd4,cd8α,cd8β,il2rβ,il2rγ,il7rα,itga4,vla1,cd49a,itga4,ia4,cd49d,itga6,vla-6,cd49f,itgad,cd11d,itgae,cd103,itgal,cd11a,lfa-1,itgam,cd11b,itgax,cd11c,itgb1,cd29,itgb2,cd18,lfa-1,itgb7,tnfr2,trance/rankl,dnam1(cd226),slamf4(cd244、2b4),cd84,cd96(tactile),ceacam1,crtam,ly9(cd229),cd160(by55),psgl1,cd100(sema4d),cd69,slamf6(ntb-a、ly108),slam(slamf1,cd150,ipo-3),blame(slamf8),selplg(cd162),ltbr,lat,gads,slp-76,pag/cbp,nkp44,nkp30,nkp46,nkg2d,toll样受体1(tlr1),tlr2,tlr3,tlr4,tlr5,tlr6,tlr7,tlr8,tlr9,本文描述的其它共刺激分子,其任何衍生物、变体、或片段,具有相同功能能力的共刺激分子的任何合成序列,和其任意组合。
在一个实施方式中,car的细胞内结构域包括一种或多种共刺激分子的任何部分,比如至少一个信号传导结构域,其来自cd3、cd8、cd27、cd28、icos、4-1bb、pd-1、其任何衍生物或变体、具有相同功能能力的其任何合成序列、和其任意组合。
在一个实施方式中,细胞内结构域包括cd3ζ信号传导结构域(seqidno:6、51或68)。在另一个实施方式中,细胞内结构域包括cd4cd28ζ(seqidno:7或62)。在另一个实施方式中,细胞内结构域包括cd28共刺激结构域(seqidno:9或67)。在又另一个实施方式中,信号传导结构域包括cd44-1bb共刺激结构域(seqidno:63)。在还另一个实施方式中,信号传导结构域包括4-1bb(cd137)共刺激结构域(seqidno:69)。在另一个实施方式中,细胞内结构域包括dap12结构域。在又另一个实施方式中,细胞内结构域包括kir结构域。细胞内结构域可以包括本文描述的一种或多种细胞内结构域。
本文描述的细胞内结构域,比如来自tcr、cd3ζ、cd3γ、cd3δ、cd3ε、cd86、普通的fcrγ、fcrβ(fcεr1b)、cd79a、cd79b、fcγriia、dap10、dap12、t细胞受体(tcr)、cd8、cd27、cd28、4-1bb(cd137)、ox9、ox40、cd30、cd40、pd-1、icos、kir家族蛋白、淋巴细胞功能相关抗原-1(lfa-1)、cd2、cd7、light、nkg2c、b7-h3、与cd83特异性地结合的配体、cds、icam-1、gitr、baffr、hvem(lightr)、slamf7、nkp80(klrf1)、cd127、cd160、cd19、cd4、cd8α、cd8β、il2rβ、il2rγ、il7rα、itga4、vla1、cd49a、itga4、ia4、cd49d、itga6、vla-6、cd49f、itgad、cd11d、itgae、cd103、itgal、cd11a、lfa-1、itgam、cd11b、itgax、cd11c、itgb1、cd29、itgb2、cd18、lfa-1、itgb7、tnfr2、trance/rankl、dnam1(cd226)、slamf4(cd244、2b4)、cd84、cd96(tactile)、ceacam1、crtam、ly9(cd229)、cd160(by55)、psgl1、cd100(sema4d)、cd69、slamf6(ntb-a、ly108)、slam(slamf1、cd150、ipo-3)、blame(slamf8)、selplg(cd162)、ltbr、lat、gads、slp-76、pag/cbp、nkp44、nkp30、nkp46、nkg2d、toll样受体1(tlr1)、tlr2、tlr3、tlr4、tlr5、tlr6、tlr7、tlr8、tlr9、或其它共刺激分子的片段或结构域,可以与本文描述的任何抗原结合结构域、本文描述的任何跨膜结构域、或可以包括在car中的本文描述的任何其它结构域组合。
在car的抗原结合结构域和跨膜结构域之间,或在car的细胞内结构域和跨膜结构域之间,可以并入间隔结构域。如本文使用的,术语“间隔结构域”通常意思是起将跨膜结构域连接至多肽链中的抗原结合结构域或细胞内结构域作用的任何寡肽或多肽。在一个实施方式中,间隔结构域可以包括多至300个氨基酸,优选地10至100个氨基酸和最优选地25至50个氨基酸。在另一个实施方式中,短的寡肽或多肽连接体,优选地长度在2和10个氨基酸之间,可以在car的跨膜结构域和细胞内结构域之间形成连接。连接体的实例包括甘氨酸-丝氨酸双联体。
人抗体
当使用car的抗原结合结构域时,可以优选地使用人抗体或其片段。完全人抗体对于人对象的治疗性处理是特别期望的。可以通过各种本领域已知的方法制造人抗体,该方法包括使用源自人免疫球蛋白序列的抗体文库的噬菌体展示方法,其包括对这些技术的改进。还参见美国专利号4,444,887和4,716,111;和pct公布号wo98/46645、wo98/50433、wo98/24893、wo98/16654、wo96/34096、wo96/33735、和wo91/10741;其每个通过引用以其全部并入本文。
还可以使用不能表达功能性内源免疫球蛋白但可以表达人免疫球蛋白基因的转基因小鼠来产生人抗体。例如,人重链和轻链免疫球蛋白基因复合体可以被随机地或通过同源重组引入小鼠胚胎干细胞。可选地,除人重链和轻链基因之外,人可变区、恒定区、和多变区也可以被引入小鼠胚胎干细胞。通过同源重组,可以与引入人免疫球蛋白基因座分开地或同时地,使小鼠重链和轻链免疫球蛋白基因非功能性。例如,已经描述了在嵌合和种系突变小鼠中纯合缺失抗体重链j区(jh)基因导致内源性抗体产生的完全抑制。修饰的胚胎干细胞被扩展并且显微注射入胚泡以产生嵌合小鼠。然后繁殖嵌合小鼠以产生表达人抗体的纯合后代。转基因小鼠以正常的方式使用选择的抗原——例如,本发明的多肽的全部或部分——进行免疫。针对选择的靶标的抗体可以使用常规的杂交瘤技术从免疫的转基因小鼠获得。由转基因小鼠聚藏(harbor)的人免疫球蛋白转基因在b细胞分化期间重排,并且随后经历类别转换和体细胞突变。因而,使用这样的技术可能产生治疗上有用的igg、iga、igm和ige抗体,包括但不限于igg1(γ1)和igg3。对于此技术用于产生人抗体的综述,参见lonberg和huszar(int.rev.immunol.,13:65-93(1995))。对于此技术用于产生人抗体和人单克隆抗体以及用于生产这样的抗体的方案的详细讨论,参见,例如,pct公布号wo98/24893、wo96/34096、和wo96/33735;和美国专利号5,413,923;5,625,126;5,633,425;5,569,825;5,661,016;5,545,806;5,814,318;和5,939,598,其每个通过引用以其全部并入本文。此外,可以与公司比如abgenix,inc.(freemont,calif.)和genpharm(sanjose,calif.)签订合同以使用与上面描述的类似的技术提供针对选择的抗原的人抗体。对于在种系突变小鼠中转移人类种系免疫球蛋白基因阵列在抗原攻击之后将导致产生人抗体的具体讨论,参见,例如,jakobovits等proc.natl.acad.sci.usa,90:2551(1993);jakobovits等nature,362:255-258(1993);bruggermann等yearinimmunol.,7:33(1993);和duchosal等nature,355:258(1992)。
人抗体也可以源自噬菌体展示文库(hoogenboom等j.mol.biol.,227:381(1991);marks等j.mol.biol.,222:581-597(1991);vaughan等naturebiotech.,14:309(1996))。噬菌体展示技术(mccafferty等nature,348:552-553(1990))可以被用于从来自未免疫供体的免疫球蛋白可变(v)结构域基因所有组成成分在体外产生人抗体和抗体片段。根据此技术,将抗体v结构域基因符合读框地克隆入丝状噬菌体比如m13或fd的主要或次要外壳蛋白基因,并且在噬菌体颗粒的表面上作为功能性抗体片段展示。因为丝状颗粒包含噬菌体基因组的单链dna拷贝,所以基于抗体的功能性质的选择也导致选择编码展示那些性质的抗体的基因。因而,噬菌体模拟了b细胞的一些性质。噬菌体展示可以以各种形式进行;对于其综述,参见,例如,johnson,kevins和chiswell,davidj.,currentopinioninstructuralbiology3:564-571(1993)。数种来源的v基因区段可以被用于噬菌体展示。clackson等nature,352:624-628(1991)从源自未免疫小鼠脾脏的v基因的小的随机组合文库分离了抗唑酮抗体的多样化阵列。可以构建来自未免疫人供体的v基因所有组成成分,并且抗原(包括自体抗原)的多样化阵列的抗体可以基本上遵循下列描述的技术进行分离:marks等j.mol.biol.,222:581-597(1991),或者griffith等emboj.,12:725-734(1993)。还参见美国专利号5,565,332和5,573,905,其每个通过引用以其全部并入本文。
人抗体也可以通过体外活化的b细胞生成(参见,美国专利号5,567,610和5,229,275,其每个通过引用以其全部并入本文)。人抗体也可以使用杂交瘤技术体外生成,比如,但不限于由roder等(methodsenzymol.,121:140-167(1986))描述的。
人源化抗体
可选地,在一些实施方式中,非人抗体可以是人源化的,其中抗体的特异性序列或区域被修饰以增加与人中天然产生的抗体的相似性。例如,在本发明中,抗体或其片段可以包括非人哺乳动物scfv。在一个实施方式中,抗原结合结构域部分是人源化的。
可以使用本领域已知的各种技术产生人源化抗体,该技术包括但不限于cdr-移植(参见,例如,欧洲专利号ep239,400;国际公布号wo91/09967;和美国专利号5,225,539、5,530,101、和5,585,089,其每个通过引用以其全部并入本文)、贴面(veneering)或表面重建(resurfacing)(参见,例如,欧洲专利号ep592,106和ep519,596;padlan,1991,molecularimmunology,28(4/5):489-498;studnicka等1994,proteinengineering,7(6):805-814;和roguska等1994,pnas,91:969-973,其每个通过引用以其全部并入本文)、链改组(参见,例如,美国专利号5,565,332,其通过引用以其全部并入本文)、和在如下中公开的技术,例如,美国专利申请公布号us2005/0042664、美国专利申请公布号us2005/0048617、美国专利号6,407,213、美国专利号5,766,886、国际公布号wo9317105、tan等j.immunol.,169:1119-25(2002)、caldas等proteineng.,13(5):353-60(2000)、morea等methods,20(3):267-79(2000)、baca等j.biol.chem.,272(16):10678-84(1997)、roguska等proteineng.,9(10):895-904(1996)、couto等cancerres.,55(23supp):5973s-5977s(1995)、couto等cancerres.,55(8):1717-22(1995)、sandhujs,gene,150(2):409-10(1994)、和pedersen等j.mol.biol.,235(3):959-73(1994),其每个通过引用以其全部并入本文。通常,框架区中的框架残基将被置换为来自cdr供体抗体的相应残基以改变,优选地改进,抗原结合。这些框架置换通过本领域熟知的方法鉴定,例如,通过建模cdr和框架残基的相互作用以鉴定对抗原结合重要的框架残基和通过序列比较鉴定特定位置处的不常见的框架残基(参见,例如,queen等,美国专利号5,585,089;和riechmann等1988,nature,332:323,其通过引用以其全部并入本文)。
人源化抗体具有从非人来源引入其的一个或多个氨基酸残基。这些非人氨基酸残基通常被称为“输入”残基,其通常取自“输入”可变结构域。因而,人源化抗体包括来自非人免疫球蛋白分子的一个或多个cdr和来自人的框架区。抗体的人源化是本领域熟知的,并且可以基本上遵循winter与合作者的方法进行(jones等nature,321:522-525(1986);riechmann等nature,332:323-327(1988);verhoeyen等science,239:1534-1536(1988)),其通过将啮齿动物cdr或cdr序列置换为相应的人抗体序列,即cdr-移植(ep239,400;pct公布号wo91/09967;和美国专利号4,816,567;6,331,415;5,225,539;5,530,101;5,585,089;6,548,640,其内容通过引用以其全部并入本文)。在这样的人源化嵌合抗体中,实质上小于完整人可变结构域已经被来自非人物种的相应的序列置换。实际上,人源化抗体通常是其中一些cdr残基和可能的一些框架(fr)残基被来自啮齿动物抗体中的类似位点的残基置换的人抗体。抗体的人源化也可以通过贴面或表面重建(ep592,106;ep519,596;padlan,1991,molecularimmunology,28(4/5):489-498;studnicka等proteinengineering,7(6):805-814(1994);和roguska等pnas,91:969-973(1994))或链改组(美国专利号5,565,332)实现,其内容通过引用以其全部并入本文。
用于制造人源化抗体的人可变结构域——轻和重二者——的选择是为了降低抗原性。根据所谓的“最佳拟合”方法,啮齿动物抗体的可变结构域的序列针对已知的人可变结构域序列的整个文库进行筛选。最接近于啮齿动物的人序列然后被接受为用于人源化抗体的人框架(fr)(sims等j.immunol.,151:2296(1993);chothia等j.mol.biol.,196:901(1987),其内容通过引用以其全部并入本文)。另一种方法使用源自特定的轻链或重链亚组的所有人抗体的共有序列的特定框架。相同的框架可以被用于数种不同的人源化抗体(carter等proc.natl.acad.sci.usa,89:4285(1992);presta等j.immunol.,151:2623(1993),其内容通过引用以其全部并入本文)。
抗体可以被人源化,并且保留对靶标抗原的高亲和力和其它有利的生物学性质。根据本发明的一个方面,通过使用亲本和人源化序列的三维模型分析亲本序列和多种概念性人源化产物的方法来制备人源化抗体。三维免疫球蛋白模型是一般可获得的并且是本领域技术人员熟悉的。图解和展示选择的候选免疫球蛋白序列的可能的三维构象结构的计算机程序是可获得的。检查这些展示允许分析残基在候选免疫球蛋白序列的功能中的可能作用,即,分析影响候选免疫球蛋白结合靶抗原的能力的残基。以此方式,fr残基可以从接受者和输入序列选择并组合,使得实现期望的抗体特性,比如对靶抗原的增加的亲和力。一般而言,cdr残基直接地和最实质性地参与影响抗原结合。
人源化抗体保留与原始抗体相似的抗原特异性。然而,使用人源化的某些方法,可以使用“定向进化”的方法增加抗体对靶标抗原的结合亲和力和/或特异性,如由wu等j.mol.biol.,294:151(1999)描述的,其内容通过引用以其全部并入本文。
载体
载体可以被用于将嵌合细胞内信号传导分子或car引入如本文其它地方描述的t细胞。在一方面,本发明包括载体,其包括编码嵌合细胞内信号传导的核酸序列。在另一方面,本发明包括载体,其包括编码car的核酸序列。在一个实施方式中,载体包括质粒载体、病毒载体、逆转录转座子(例如背负式(piggyback)、睡美人式(sleepingbeauty))、定点插入载体(例如crispr、锌指核酸酶、talen)、或自杀表达载体、或本领域其它已知的载体。
上面提及的所有构建体能够与第3代慢病毒载体质粒、其它病毒载体、或批准用于人细胞的rna一起使用。在一个实施方式中,载体是病毒载体,比如慢病毒载体。在另一个实施方式中,载体是rna载体。
可以通过测序验证本文描述的任何分子的产生。可以使用免疫印迹、免疫组织化学、流式细胞术或本领域熟知和可利用的其它技术验证全长蛋白质的表达。
本发明也提供了其中插入本发明的dna的载体。载体,包括源于逆转录病毒比如慢病毒的那些,是实现长期基因转移的合适工具,这是由于它们允许转基因的长期、稳定整合并且其在子细胞中增殖。慢病毒载体具有超过源自致癌逆转录病毒比如鼠白血病病毒的载体的额外优点,因为它们可转导非增殖的细胞,比如肝细胞。它们也具有在将其引入的对象中导致低免疫原性的额外优点。
通常通过可操作地连接核酸或其部分至启动子,并且将构建体并入表达载体来实现天然或合成核酸的表达。载体是通常能够在哺乳动物细胞中复制和/或还能够整合入哺乳动物的细胞基因组的载体。典型的载体包含对调节期望的核酸序列的表达有用的转录和翻译终止子、初始序列、和启动子。
核酸可以被克隆入任何数目的不同类型的载体。例如,核酸可以被克隆入如下载体:其包括但不限于质粒、噬菌粒、噬菌体衍生物、动物病毒、和粘粒。特别感兴趣的载体包括表达载体、复制载体、探针生成载体、和测序载体。
表达载体可以以病毒载体的形式被提供给细胞。病毒载体技术是本领域熟知的并且例如在sambrook等2012,molecularcloning:alaboratorymanual,volumes1-4,coldspringharborpress,ny和其它病毒学和分子生物学手册中描述。可用作载体的病毒包括但不限于逆转录病毒、腺病毒、腺伴随病毒、疱疹病毒、和慢病毒。一般而言,合适的载体包含在至少一种生物体中起作用的复制起点、启动子序列、方便的限制酶位点、和一个或多个可选择的标志物(例如,wo01/96584;wo01/29058;和美国专利号6,326,193)。
额外的启动子元件,例如增强子,调控转录起始的频率。通常,这些位于起始位点上游30-110bp的区域,但是最近已经显示许多启动子也包含起始位点下游的功能元件。启动子元件之间的间隔经常是柔性的,使得当元件相对于另一个被倒置或移动时保持启动子功能。在胸苷激酶(tk)启动子中,启动子元件之间的间隔可被增加隔开50bp,然后活性才开始下降。取决于启动子,显示出单个元件可以合作地或独立地起作用以起动转录。
启动子的实例是即时早期巨细胞病毒(cmv)启动子序列。此启动子序列是能够驱动可操作地连接至其的任何多核苷酸序列的高水平表达的强的组成型启动子序列。然而,也可以使用其它组成型启动子序列,包括但不限于猿猴病毒40(sv40)早期启动子、小鼠乳癌病毒(mmtv)、人免疫缺陷病毒(hiv)长末端重复(ltr)启动子、momulv启动子、鸟类白血病病毒启动子、eb病毒即时早期启动子、鲁斯氏肉瘤病毒启动子、ef-1α启动子,以及人基因启动子,比如,但不限于肌动蛋白启动子、肌球蛋白启动子、血红素启动子、和肌酸激酶启动子。进一步,本发明应当不限于使用组成型启动子。诱导型启动子也被考虑作为本发明的一部分。诱导型启动子的使用提供了分子开关,其能够当期望这样的表达时,开启可操作地连接诱导型启动子的多核苷酸序列的表达,或当不期望表达时关闭表达。诱导型启动子的例子包括但不限于金属硫蛋白启动子、糖皮质激素启动子、孕酮启动子、和四环素启动子。
为了评估多肽或其部分的表达,引入细胞的表达载体也可以包含可选择的标记物基因或报道基因中的任一种或二者,以促进从通过病毒载体寻求被转染或感染的细胞群鉴定和选择表达细胞。在其它方面,可选择的标志物可以被携带在dna的单独段上并且用于共转染程序。可选择的标志物和报道基因二者均可以侧接适当的调控序列,以使得能够在宿主细胞中表达。有用的可选择的标志物包括例如抗生素抗性基因,比如neo等。
报道基因被用于鉴定潜在转染的细胞并且用于评价调控序列的功能性。一般而言,报道基因是如下基因:其不存在于受体生物体或组织中或由受体生物体或组织表达,并且编码多肽,该多肽的表达由一些可容易检测的性质例如酶促活性显现。在dna已经被引入受体细胞后,在合适的时间评估报道基因的表达。合适的报道基因可以包括编码萤光素酶、β-半乳糖苷酶、氯霉素乙酰转移酶、分泌型碱性磷酸酶的基因,或绿色荧光蛋白基因(例如,ui-tei等2000febsletters479:79-82)。合适的表达系统是熟知的并且可以使用已知的技术制备或商购。一般而言,显示最高水平的报道基因表达的具有最小5’侧翼区的构建体被鉴定为启动子。这样的启动子区可以被连接至报道基因并且被用于评价试剂调节启动子驱动的转录的能力。
引入核酸
将基因——比如嵌合细胞内信号传导分子或car——引入细胞和表达该基因的方法是本领域已知的。在表达载体的上下文中,可以通过本领域的任何方法将载体容易地引入宿主细胞,例如,哺乳动物、细菌、酵母、或昆虫细胞。例如,表达载体可以通过物理、化学、或生物学手段被转移入宿主细胞。
用于将多核苷酸引入宿主细胞的物理方法包括磷酸钙沉淀、脂质转染、粒子轰击、显微注射、电穿孔等。用于产生包括载体和/或外源核酸的细胞的方法是本领域熟知的。参见,例如,sambrook等2012,molecularcloning:alaboratorymanual,volumes1-4,coldspringharborpress,ny。可以使用商业上可获得的方法将核酸引入靶标细胞,该方法包括电穿孔(amaxanucleofector-ii(amaxabiosystems,cologne,germany))、ecm830(btx)(harvardinstruments,boston,mass.)或genepulserii(biorad,denver,colo.)、multiporator(eppendort,hamburggermany)。还可以通过如下将核酸引入细胞:使用阳离子脂质体介导的转染、使用脂质转染、使用聚合物封装、使用肽介导的转染、或使用生物射弹粒子递送系统比如“基因枪”(参见,例如,nishikawa等humgenether.,12(8):861-70(2001)。
用于将感兴趣的多核苷酸引入宿主细胞的生物学方法包括使用dna和rna载体。rna载体包括具有rna启动子和/或用于产生rna转录物的其它相关结构域的载体。病毒载体,并且尤其是逆转录病毒载体,已经成为最广泛使用的将基因插入哺乳动物例如人细胞的方法。其它病毒载体可以源自慢病毒、痘病毒、单纯疱疹病毒、腺病毒和腺伴随病毒等。参见,例如,美国专利号5,350,674和5,585,362。
用于将多核苷酸引入宿主细胞的化学手段包括胶体分散系统,比如大分子复合物、纳米胶囊、微球、珠和基于脂质的系统,包括水包油乳液、胶团、混合胶团、和脂质体。用作体外和体内递送媒介的示例性胶体系统是脂质体(例如,人工膜囊)。
在利用非病毒递送系统的情况下,示例性递送媒介是脂质体。考虑将脂质制剂用于将核酸引入宿主细胞(体外、离体或体内)。在另一方面,核酸可以与脂质缔合。与脂质缔合的核酸可以被封装在脂质体的水性内部中,散布在脂质体的脂双层内,经由与脂质体和寡核苷酸二者均缔合的连接分子附加至脂质体,包埋在脂质体中,与脂质体复合,分散在包含脂质的溶液中,与脂质混合,与脂质组合,作为悬浮液包含在脂质中,包含在胶团中或与胶团复合,或以其它方式与脂质缔合。脂质、脂质/dna或脂质/表达载体缔合的组合物不限于溶液中的任何具体结构。例如,它们可以存在于双分子层结构中,作为胶团,或具有“坍缩的(collapsed)”结构。它们也可简单地被散布在溶液中,可能形成大小或形状不均一的聚集体。脂质是脂肪物质,其可以是天然存在的或合成的脂质。例如,脂质包括脂肪液滴——其在细胞质中天然存在——以及包含长链脂肪族烃和它们的衍生物比如脂肪酸、醇类、胺类、氨基醇类、和醛类的化合物类别。
适于使用的脂质可以从商业来源获得。例如,二肉豆蔻酰磷脂酰胆碱(“dmpc”)可以从sigma,st.louis,mo获得;磷酸二鲸蜡酯(“dcp”)可以从k&klaboratories(plainview,ny)获得;胆固醇(“choi”)可以从calbiochem-behring获得;二肉豆蔻酰磷脂酰甘油(“dmpg”)和其它脂质可以从avantipolarlipids,inc.(birmingham,al.)获得。氯仿或氯仿/甲醇中的脂质原液可以被储存在大约-20℃下。氯仿被用作唯一的溶剂,这是由于它比甲醇更容易蒸发。“脂质体”是通用术语,其涵盖通过生成封闭的脂双层或聚集体而形成的各种单一和多层脂质媒介。脂质体可以被表征为具有含有磷脂双层膜和内部水性介质的囊泡结构。多层脂质体具有由水性介质分隔的多个脂质层。当磷脂悬浮在过量的水性溶液中时,它们自发地形成。在形成封闭结构和在脂双层之间包埋水和溶解的溶质前,脂质组分经历自我重排(ghosh等1991glycobiology5:505-10)。然而,也涵盖与正常的囊泡结构相比在溶液中具有不同结构的组合物。例如,脂质可以呈现胶团结构或仅作为脂质分子的非均一聚集体存在。同样考虑脂质转染胺-核酸复合物。
不管用于将外源核酸引入宿主细胞或以其它方式将细胞暴露于本文描述的分子的方法,为了确认宿主细胞中核酸的存在,可以进行各种试验。这样的试验包括例如本领域技术人员熟知的“分子生物学”试验,比如dna印迹和rna印迹、rt-pcr和pcr;“生物化学”试验,比如例如通过免疫学手段(ellsa和蛋白质印迹)或通过本文描述的鉴定落入本发明范围内的试剂的试验来检测特定肽的存在或不存在。
在一个实施方式中,通过选自如下的方法引入在本文其它地方描述的核酸序列中的一种或多种:转导细胞群、转染细胞群、和电穿孔细胞群。在一个实施方式中,细胞群包括本文描述的核酸序列中的一种或多种。
在一个实施方式中,引入细胞的核酸是rna。在另一个实施方式中,rna是包括体外转录的rna或合成rna的mrna。使用聚合酶链式反应(pcr)生成的模板通过体外转录产生rna。来自任何来源的感兴趣的dna可以通过pcr直接转化为使用适当的引物和rna聚合酶的体外mrna合成的模板。dna的来源可以是,例如,基因组dna、质粒dna、噬菌体dna、cdna、合成dna序列或任何其它适当的dna来源。期望的体外转录模板是嵌合细胞内信号传导分子。
pcr可以被用于生成体外转录mrna——其随后被引入细胞——的模板。用于进行pcr的方法是本领域熟知的。用于pcr的引物被设计为具有与待用作pcr模板的dna的区域基本上互补的区域。如本文使用的“基本上互补的”指的是如此核苷酸的序列:其中引物序列中的大部分或所有碱基是互补的,或一个或多个碱基是非互补的或错配的。基本上互补的序列能够与目标dna靶标在用于pcr的退火条件下退火或杂交。引物可以被设计为与dna模板的任何部分基本上互补。例如,引物可以被设计为扩增在细胞中正常转录的部分基因(开放阅读框),包括5'和3'utr。引物还可以被设计为扩增编码感兴趣的特定结构域的部分基因。在一个实施方式中,引物被设计为扩增包括所有或部分5'和3'utr的人cdna的编码区。通过本领域熟知的合成方法生成对pcr有用的引物。“正向引物”是包含与待扩增的dna序列上游的dna模板上的核苷酸基本上互补的核苷酸区域的引物。本文使用的“上游”指的是待扩增的dna序列相对于编码链的5'位置。“反向引物”是包含与待扩增的dna序列下游的双链dna模板基本上互补的核苷酸区域的引物。本文使用的“下游”指的是待扩增的dna序列相对于编码链的3'位置。
还可以使用具有促进rna的稳定性和/或翻译效率的能力的化学结构。rna优选地具有5'和3'utr。在一个实施方式中,5'utr的长度在零和3000个核苷酸之间。待添加至编码区的5'和3'utr序列的长度可以通过不同的方法改变,包括但不限于设计用于pcr的引物,其退火至utr的不同区域。使用此方法,本领域普通技术人员可以修改实现转染转录的rna之后最佳翻译效率所需要的5'和3'utr长度。
5'和3'utr可以是感兴趣的基因的天然存在的内源性5'和3'utr。可选地,可以通过将utr序列并入正向和反向引物或通过模板的任何其它修饰添加对感兴趣的基因非内源性的utr序列。使用对感兴趣的基因非内源性的utr序列对修改rna的稳定性和/或翻译效率可以是有用的。例如,已知3'utr序列中的富au元件可以降低mrna的稳定性。因此,3'utr可以被选择或设计以基于本领域熟知的utr的性质增加转录的rna的稳定性。
在一个实施方式中,5'utr可以包含内源基因的kozak序列。可选地,当正在通过如上面描述的pcr添加对感兴趣的基因非内源性的5'utr时,可以通过添加5'utr序列重新设计共有的kozak序列。kozak序列可以增加一些rna转录物的翻译效率,但是似乎不是所有rna能够高效翻译所需要的。许多mrna的kozak序列的要求是本领域已知的。在其它实施方式中,5'utr可以源自其rna基因组在细胞中稳定的rna病毒。在其它实施方式中,多种核苷酸类似物可以被用于3'或5'utr以阻碍mrna的核酸外切酶降解。
为了使得能够从dna模板合成rna而不需要基因克隆,转录的启动子应当被附加至待转录的序列上游的dna模板。当作为rna聚合酶的启动子起作用的序列被添加至正向引物的5'端时,rna聚合酶启动子并入待转录的开放阅读框上游的pcr产物。在一个实施方式中,启动子是t7聚合酶启动子,如本文其它地方描述的。其它有用的启动子包括但不限于t3和sp6rna聚合酶启动子。t7、t3和sp6启动子的共有的核苷酸序列是本领域已知的。
在一个实施方式中,mrna具有5'端上的帽和3'聚腺苷酸尾二者,其决定核糖体结合、翻译起始和mrna在细胞中的稳定性。在环状dna模板例如质粒dna上,rna聚合酶产生不适于在真核细胞中表达的长的多联体产物(concatamericproduct)。在3'utr的端处线性化的质粒dna的转录产生正常大小的mrna,其在真核转染中无效,即使其在转录后被聚腺苷酸化。
在线性dna模板上,噬菌体t7rna聚合酶可以使转录物的3'端延伸超过模板的最后一个碱基(schenborn和mierendorf,nucacidsres.,13:6223-36(1985);nacheva和berzal-herranz,eur.j.biochem.,270:1485-65(2003))。
将聚a/t区段整合入dna模板的常规方法是分子克隆。然而,整合入质粒dna的聚a/t序列可能引起质粒不稳定,这就是为什么从细菌细胞获得的质粒dna模板经常被缺失和其它畸变高度污染。这使得克隆程序不仅费力而且耗时,并且经常不可靠。这就是为什么允许构建具有聚a/t3'区段的dna模板而没有克隆的方法是高度期望的。
转录的dna模板的聚a/t区段可以在pcr期间通过使用包含聚t尾——比如100个t尾(大小可以是50-5000个t)——的反向引物产生,或在pcr后通过任何其它方法产生,该方法包括但不限于dna连接或体外重组。聚腺苷酸尾还给rna提供稳定性并且减少它们的降解。通常,聚腺苷酸尾的长度与转录的rna的稳定性正相关。在一个实施方式中,聚腺苷酸尾在100和5000个腺苷之间。
可以在体外转录之后使用聚腺苷酸聚合酶——比如大肠杆菌聚腺苷酸聚合酶(e-pap)——进一步延伸rna的聚腺苷酸尾。在一个实施方式中,使聚腺苷酸尾的长度从100个核苷酸增加至300和400之间个核苷酸导致rna的翻译效率增加大约两倍。另外,不同的化学基团附加至3'端可以增加mrna稳定性。这样的附加可以包含修饰的/人工的核苷酸、适配体和其它化合物。例如,atp类似物可以使用聚腺苷酸聚合酶并入聚腺苷酸尾。atp类似物可以进一步增加rna的稳定性。
5'帽也给rna分子提供稳定性。在优选的实施方式中,通过本文公开的方法产生的rna包括5'帽。使用本领域已知的和本文描述的技术提供5'帽(cougot等trendsinbiochem.sci.,29:436-444(2001);stepinski等rna,7:1468-95(2001);elango等biochim.biophys.res.commun.,330:958-966(2005))。
通过本文公开的方法产生的rna还可以包含内部核糖体进入位点(ires)序列。ires序列可以是起始帽-独立性核糖体结合至mrna并且促进翻译起始的任何病毒序列、染色体序列或人工设计的序列。可以包括适于细胞电穿孔的任何溶质,其可以包含促进细胞通透性和生存力的因子,比如糖、肽、脂质、蛋白质、抗氧化剂、和表面活性剂。
一些体外转录的rna(ivt-rna)载体是文献中已知的,其以标准化方式被用作体外转录的模板并且已经以如此方式被基因修饰:该方式产生稳定的rna转录物。用于本领域的目前的方案基于具有下列结构的质粒载体:使得能够rna转录的5'rna聚合酶启动子,接着是由非翻译区域(utr)在3'和/或5'侧接的感兴趣的基因,和包含50-70个a核苷酸的3'聚腺嘌呤基盒。在体外转录之前,环状质粒在聚腺嘌呤基盒下游通过ii型限制酶(识别序列对应于切割位点)线性化。聚腺嘌呤基盒因而对应于转录物中的后面的聚腺苷酸序列。作为此程序的结果,一些核苷酸在线性化后仍作为部分酶切割位点保留并且延伸或掩盖3'端处的聚腺苷酸序列。尚不清楚此非生理学突出部分是否影响从这样的构建体细胞内产生的蛋白质的量。
在一方面,rna构建体通过电穿孔被递送入细胞。参见,例如,如在us2004/0014645、us2005/0052630a1、us2005/0070841a1、us2004/0059285a1、us2004/0092907a1中教导的将核酸构建体电穿孔入哺乳动物细胞的制剂和方法。包括电穿孔任何已知的细胞类型所需要的电场强度的多个参数在相关的研究文献以及领域的众多专利和申请中通常是已知的。参见例如,美国专利号6,678,556、美国专利号7,171,264、和美国专利号7,173,116。用于电穿孔的治疗性应用的设备是商业上可获得的,例如,medpulsertmdna电穿孔治疗系统(inovio/genetronics,sandiego,calif.),并且在专利比如美国专利号6,567,694;美国专利号6,516,223、美国专利号5,993,434、美国专利号6,181,964、美国专利号6,241,701、和美国专利号6,233,482中描述;电穿孔也可以被用于细胞体外转染,例如,如在us20070128708a1中描述的。电穿孔也可以被用于将核酸体外递送入细胞。因此,利用本领域技术人员已知的许多可用的装置和电穿孔系统中的任一种将包括核酸的表达构建体电穿孔介导的施用入细胞提供了令人激动的将感兴趣的rna递送至靶标细胞的新手段。
可选地,在另一方面,本发明包括用于生成修饰的t细胞的方法,其包括使用编码嵌合细胞内信号传导分子的核酸序列电穿孔t细胞群,其中核酸序列包括共刺激分子的细胞内结构域的核酸序列并且基本上缺少细胞外结构域。在一个实施方式中,编码嵌合细胞内信号传导分子的核酸序列被电穿孔入细胞。在又另一个实施方式中,编码car或膜结合嵌合受体的核酸序列被进一步电穿孔入细胞。
t细胞的来源
修饰的t细胞可以由任何来源的t细胞生成。在一个实施方式中,t细胞的来源从对象获得。对象的非限制性实例包括人、狗、猫、小鼠、大鼠、和其转基因物种。优选地,对象是人。t细胞可以从许多来源获得,包括外周血单核细胞、骨髓、淋巴结组织、脾脏组织、脐带、和肿瘤。在某些实施方式中,可以使用本领域中可利用的任意数目的t细胞系。在某些实施方式中,可以使用技术人员已知的任意数目的技术比如ficoll分离从由对象收集的单位血液获得t细胞。在一个实施方式中,通过单采血液成分术或白细胞提取法获得来自个体的循环血液的细胞。单采血液成分术产物通常包含淋巴细胞,包括t细胞、单核细胞、粒细胞、b细胞、其它成核的白细胞、红细胞、和血小板。可以洗涤通过单采血液成分术收集的细胞以去除血浆部分并且将细胞放置在适当的缓冲液或介质中,用于后续处理步骤,该缓冲液或介质比如磷酸盐缓冲盐水(pbs)或洗涤液,其缺少钙并且可能缺少镁或可能缺少很多——如果不是全部的话——二价阳离子。在洗涤后,细胞可以被重悬在各种生物相容性缓冲液中,比如,例如,无ca、无mgpbs。可选地,可以去除单采血液成分术样品的非期望组分,并且将细胞直接重悬在培养基中。
在另一个实施方式中,通过溶解红细胞和耗尽单核细胞从外周血分离t细胞,例如,通过经由percolltm梯度的离心。可选地,可以从脐带分离t细胞。无论如何,可以通过正或负选择技术进一步分离t细胞的特定亚群。
如此分离的脐带血单核细胞可以被耗尽表达某些抗原——包括但不限于cd34、cd8、cd14、cd19和cd56——的细胞。可以使用分离的抗体、包括抗体的生物学样品比如腹水、结合至物理支持体(support)的抗体、和细胞结合的抗体完成这些细胞的耗尽。
可以使用针对对负选择的细胞独特的表面标志物的抗体的组合完成通过负选择富集t细胞群。优选的方法是细胞分选和/或选择,其经由负磁性免疫粘附或使用针对在负选择的细胞上存在的细胞表面标志物的单克隆抗体的混合物的流式细胞术。例如,为了通过负选择富集cd4+细胞,单克隆抗体混合物通常包括cd14、cd20、cd11b、cd16、hla-dr、和cd8的抗体。
对于通过正或负选择分离期望的细胞群,可以改变细胞和表面(例如,颗粒比如珠)的浓度。在某些实施方式中,可能期望的是,显著地降低珠和细胞被混合在一起的体积(即,增加细胞的浓度),以确保细胞和珠的最大接触。例如,在一个实施方式中,使用20亿个细胞/ml的浓度。在一个实施方式中,使用10亿个细胞/ml的浓度。在进一步的实施方式中,使用大于1亿个细胞/ml。在进一步的实施方式中,使用10、15、20、25、30、35、40、45、或50×106个细胞/ml的细胞浓度。在又另一个实施方式中,使用75、80、85、90、95、或100×106个细胞/ml的细胞浓度。在进一步的实施方式中,可以使用125或150×106个细胞/ml的浓度。使用高浓度可以产生增加的细胞产率、细胞活化、和细胞扩展。
在洗涤步骤后,还可以冷冻t细胞,其不需要单核细胞-去除步骤。虽然不希望被理论束缚,但是通过去除细胞群中的粒细胞和一定程度的单核细胞,冷冻和后续的解冻步骤提供了更均一的产物。在去除血浆和血小板的洗涤步骤后,细胞可以被悬浮在冷冻液中。虽然许多冷冻液和参数是本领域已知的并且将在此背景下是有用的,但是在非限制性实例中,一种方法涉及使用包含20%dmso和8%人血清白蛋白的pbs,或其它合适的细胞冷冻介质,细胞然后以1°/分钟的速率被冷冻至-80℃,并且储存在液氮储罐的蒸汽相中。可以使用受控冷冻的其它方法以及在-20℃下或在液氮中不受控的即刻冷冻。
在一个实施方式中,细胞群包括本发明的t细胞。细胞群的实例包括但不限于外周血单核细胞、脐带血细胞、纯化的t细胞群、和t细胞系。在另一个实施方式中,外周血单核细胞包括t细胞群。在又另一个实施方式中,纯化的t细胞包括t细胞群。
t细胞的扩展
可以离体扩展通过本文描述的任何方法生成的t细胞。在一个实施方式中,培养t细胞或包括t细胞的细胞群进行扩展。通常,t细胞通过与表面接触被扩展,该表面具有附加至其的刺激cd3/tcr复合体相关信号的试剂和刺激t细胞表面上的共刺激分子的配体。
本文描述了用于扩展t细胞的方法,例如,t细胞可以被扩展大约10倍、20倍、30倍、40倍、50倍、60倍、70倍、80倍、90倍、100倍、200倍、300倍、400倍、500倍、600倍、700倍、800倍、900倍、1000倍、2000倍、3000倍、4000倍、5000倍、6000倍、7000倍、8000倍、9000倍、10,000倍、100,000倍、1,000,000倍、10,000,000倍、或更大,和其间的任何和所有全部或部分整数。在一个实施方式中,t细胞以大约20倍至大约50倍的范围扩展。
在将细胞传代至另一个培养设备前,t细胞可以在培养设备中的细胞培养基中培育一段时间或直到细胞达到进行最佳传代的汇合或高细胞密度。培养设备可以具有一般用于体外培养细胞的任何培养设备。优选地,在将细胞传代至另一个培养设备前,汇合水平是70%或更大。更优选地,汇合水平是90%或更大。一段时间可以是适于细胞体外培养的任何时间。可以在t细胞培养的任何时间替换t细胞培养基。优选地,大约每2至3天替换t细胞培养基。然后从培养设备采集t细胞,于是t细胞可以被立即使用或冷藏储存以备后用。在一个实施方式中,本发明包括冷藏扩展的t细胞。在将在本文其它地方描述的分子中的一种或多种引入t细胞之前解冻冷藏的t细胞。
如本文描述的培养步骤(与如本文描述的试剂接触)可以非常短,例如小于24小时比如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、或23小时。如本文进一步描述的培养步骤(与如本文描述的试剂接触)可以较长,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、或更多天。
在一个实施方式中,t细胞可以被培养数小时(大约3小时)至大约14天或其间的任何小时的整数值。适于t细胞培养的条件包括适当的培养基(例如,最小必需培养基或rpmi培养基1640或x-vivo15,(lonza)),其可以包含增殖和生存力必需的因子,包括血清(例如,胎牛血清或人血清)、白细胞介素-2(il-2)、胰岛素、ifn-γ、il-4、il-7、gm-csf、il-10、il-12、il-15、tgf-β、和tnf-α、或技术人员已知的用于细胞生长的任何其它添加剂。用于细胞生长的其它添加剂包括但不限于表面活性剂、人血浆蛋白粉(plasmanate)、和还原剂比如n-乙酰-半胱氨酸和2-巯基乙醇。培养基可以包括rpmi1640、aim-v、dmem、mem、α-mem、f-12、x-vivo15、和x-vivo20、优化物(optimizer)(具有添加的氨基酸、丙酮酸钠、和维生素)、无血清或补充有适当量的血清(或血浆)或限定的激素组、和/或足够t细胞的生长和扩展的量的细胞因子(一种或多种)。抗生素,例如,青霉素和链霉素,仅包括在实验培养物中,而不在被注入对象的细胞培养物中。靶标细胞被维持在支持生长必需的条件下,例如,适当的温度(例如,37℃)和气氛(例如,空气加5%co2)。
t细胞培养基可以包括可以共刺激t细胞的试剂。例如,可以刺激cd3的试剂是cd3的抗体,并且可以刺激cd28的试剂是cd28的抗体。这是因为,如由本文公开的数据展现的,通过本文公开的方法分离的细胞可以被扩展大约10倍、20倍、30倍、40倍、50倍、60倍、70倍、80倍、90倍、100倍、200倍、300倍、400倍、500倍、600倍、700倍、800倍、900倍、1000倍、2000倍、3000倍、4000倍、5000倍、6000倍、7000倍、8000倍、9000倍、10,000倍、100,000倍、1,000,000倍、10,000,000倍、或更大。在一个实施方式中,通过培养电穿孔的群体以大约20倍至大约50倍或更多的范围扩展t细胞。
疗法
在一方面,本发明包括治疗对象中与hiv感染相关联的疾病或病症的方法,其包括给对象施用治疗有效量的包括本文描述的修饰的t细胞的药物组合物。
如本文描述的修饰的t细胞可以被施用至动物,优选地哺乳动物,甚至更优选地人,以治疗hiv感染。此外,本发明的修饰的t细胞可以被用于任何病症的治疗,其中减弱的或另外抑制的免疫应答,尤其是细胞介导的免疫应答,期望地治疗或减轻疾病。
本发明的修饰的t细胞的施用可以以本领域技术人员已知的任何常规方式实施。本发明的修饰的t细胞可以通过雾化吸入、注射、吞咽、输注、植入或移植被施用至对象。本文描述的组合物可以经动脉、皮下、皮内、瘤内、结内、髓内、肌肉内、通过静脉内(i.v.)注射、或腹膜内施用至患者。在其它情况下,本发明的修饰的t细胞被直接注入对象中的炎症部位、对象中的局部疾病部位、淋巴结、器官、肿瘤等。
药物组合物
本发明的药物组合物可以包括如本文描述的修饰的t细胞,其与一种或多种药学上或生理学上可接受运载体、稀释剂或赋形剂组合。这样的组合物可以包括缓冲液比如中性缓冲盐水、磷酸盐缓冲盐水等;碳水化合物比如葡萄糖、甘露糖、蔗糖或葡聚糖、甘露醇;蛋白质;多肽或氨基酸比如甘氨酸;抗氧化剂;螯合剂比如edta或谷胱甘肽;佐剂(例如,氢氧化铝);和防腐剂。本发明的组合物优选地配制用于静脉内施用。
本发明的药物组合物可以以适于待治疗(或预防)的疾病的方式施用。施用数量和频率将通过如下因素确定,比如患者的状况以及患者疾病的类型和严重度,但是可以通过临床试验确定适当的剂量。
当指示“免疫有效量”、“抗免疫应答有效量”、“免疫应答-抑制有效量”、或“治疗量”时,可以由医师考虑个体年龄、重量、免疫应答、和患者(对象)的状况的差异确定待施用的本发明的组合物的精确量。通常可以规定可以以104至109个细胞/kg体重,优选地105至106个细胞/kg体重的剂量——包括那些范围内的所有整数值——施用包括本文描述的t细胞的药物组合物。也可以以这些剂量多次施用t细胞组合物。可以通过使用免疫疗法中公知的输注技术(参见,例如,rosenberg等neweng.j.ofmed.319:1676,1988)施用细胞。特定患者的最佳剂量和治疗方案可以通过监测患者的疾病迹象和因此调节治疗由医学领域技术人员容易地确定。
在某些实施方式中,可以期望给对象施用修饰的t细胞,并且随后重新抽取(redraw)血液(或进行单采血液成分术),根据本发明在代谢上增强来自其的t细胞,并且使用这些修饰的t细胞再输注患者。此过程可以每几周实施多次。在某些实施方式中,可以从大约10ml至大约400ml的血液抽取物获得t细胞。在某些实施方式中,从大约20ml、30ml、40ml、50ml、60ml、70ml、80ml、90ml、或100ml的血液抽取物获得修饰的t细胞。不被理论束缚,使用此多重血液抽取/多重再输注方案,可以选择出某些t细胞群。
在本发明的某些实施方式中,使用本文描述的方法修饰t细胞,并且使用本文描述的方法或本领域已知的其它方法刺激、活化或扩展t细胞,其中t细胞被扩展至治疗水平,其连同任意数目的相关治疗模态(例如,之前、同时或之后)被施用至患者,该治疗模态包括但不限于任何可用的抗-hiv疗法。
施用给患者的上面的治疗的剂量将随着正在治疗的病症的精确性质和治疗的接受者变化。应当理解在本发明中有用的方法和组合物不限于在实施例中陈述的具体制剂。下列实施例被提出以便于为本领域普通技术人员提供如何制造和使用本发明的细胞、扩展和培养方法以及治疗方法的完整的公开内容和描述,并且不意图限制本发明人视为其发明的范围。
除非另外指示,本发明的实践采用分子生物学(包括重组技术)、微生物学、细胞生物学、生物化学和免疫学的常规技术,其非常在技术人员的见识内。这样的技术在如下文献中充分地说明,比如,“molecularcloning:alaboratorymanual”,fourthedition(sambrook,2012);“oligonucleotidesynthesis”(gait,1984);“cultureofanimalcells”(freshney,2010);“methodsinenzymology”“handbookofexperimentalimmunology”(weir,1997);“genetransfervectorsformammaliancells”(miller和calos,1987);“shortprotocolsinmolecularbiology”(ausubel,2002);“polymerasechainreaction:principles,applicationsandtroubleshooting”,(babar,2011);“currentprotocolsinimmunology”(coligan,2002)。这些技术适用于产生本发明的多核苷酸和多肽,并且正因如此,可以考虑用于完成和实践本发明。将在下列部分中讨论具体实施方式的特别有用的技术。
实验实施例
通过参考下列实验实施例进一步详细地描述本发明。除非另外规定,这些实施例仅出于说明目的提供,并且不意图是限制性的。因而,本发明决不应当解释为限于下列实施例,而是应当解释为涵盖由于本文提供的教导而变得明显的任何和所有的变型。
在没有进一步描述的情况下,认为使用在先的描述和下列说明性实施例,本领域普通技术人员可以制造和利用本发明的化合物,并且实践要求保护的方法。因此,下列工作实施例具体地指出本发明的优选的实施方式,并且不解释为以任何方式限制本公开内容的其余部分。
现在描述在这些实验中采用的材料和方法。
基于单链可变片段(scfv)的car构建体:
scfv使用cd137和cd3ζ信号传导结构域合成为第2代car中的单个转录物或借助kirs2连同dap12合成为人工nk细胞受体。所有构建体遵循专利权密码子改装指数(geneart,lifetechn.)被密码子优化,用于在人中表达为一个转录物。
已经在本发明中产生和测试了下列构建体:
靶向v1/v2聚糖的“pgdm1400”(seqidno:10-13),靶向v3/v4聚糖的“pgt128”(seqidno:14-17),靶向cd4结合位点的“vrc01”(seqidno:18-21和34-35),靶向cd4结合位点的“vrc01c-mut”(hcdr1和hcdr3之间非典型的二硫键的突变)(seqidno:26-29),靶向cd4结合位点的“3bnc60”(seqidno:22-25),和cd4dap12-kirs2(seqidno:30-31)。
载体构建:
获得原始临床试验载体的骨架prt43.2gfp(liu和eiden.retrovirology9,51(2012))并且限制位点连接体被插入psti和sali位点,去除cmv启动子区。在pgk启动子下表达的cd4ζ序列由质粒prrl.pgk.f3——其具有寡核苷酸
5’gtatcgatcacgagactagc(seqidno:40)和
5’ttaaaccggtgtctggcctttgagtggtga(seqidno:41)——扩增并且被插入prt43.2内的连接体中的xhoi和agei位点。通过扩增产生ptrpecd4ζ。使用下列引物通过pcr由prrl.pgk.f3逆转录病毒骨架——其包含原始临床试验cd4-cd3ζ构建体——扩增cd4细胞外结构域:
引物-1有义(seqidno:36):
5'-ttaatgggatccatgaaccggggagtcccttt-3'
引物-2反义(seqidno:37):
5'-aaggacttccggatggctgcaccggggtggaccatg-3'
pcr产物然后被插入pelns慢病毒骨架——其包含cd8α细胞外铰链和跨膜结构域以及4-1bb和cd3ζ细胞内共刺激结构域(icd)——中的bamhi和bspe1限制位点。获得包含cd8α铰链-cd8αtm-cd3ζ或包含cd8α铰链-cd28tm-cd28-cd3ζicd的pelns慢病毒载体并且被用作模板以使用下列引物pcr扩增铰链-tm-和icd区:
引物-3有义(seqidno:38):
5’-gggacactccggaaccacgacgccagcgccgcg-3’
引物-4反义(seqidno:39):
5’-gggacacgtcgacttagcgagggggca-3’
此pcr模板然后被插入最近生成的pelnscd44-1bbζ构建体中的bspe1和sal1位点以替换包含cd8α铰链-4-1bb-cd3ζ区的区。质粒然后被电穿孔入大肠杆菌的top10菌株并且在其中繁殖。
通过合成tcrα和tcrβ基因序列(idt)生成表达能够识别hivp24gag表位kafspevipmf(seqidno:42,ptrpeb57-kf11)的b57局限的tcr的慢病毒载体。如先前描述的,tcrα和tcrβ基因序列被t2a分隔,其允许两种tcr基因的协调表达(varela-rohena等naturemedicine14,1390-1395(2008))。vrc01、3bnc60、pgt128、和pgdm1400scfvcar由公开的亲本抗体序列以轻链-连接体-重链构型生成。连接体序列如下:ggssrssssggggsgggg(seqidno:43)。氨基酸序列被密码子优化(geneart)并且被合成为双链dna片段(idt或geneart),侧接合适的限制位点并且然后被克隆入具有bamhi和bspe1限制位点的ptrpe表达质粒。pg9scfv被克隆入具有bamhi和bspe1限制位点的ptrpe表达质粒。用于此研究的优化的hivcar的序列中的一些在图18(seqidno:44-61,氨基酸)和图19(seqidno:62-79,核酸)中列出。
基于vrc01、vrc01c-、和3bnc60scfv的kir和igg4-41bb-ζcar的构建。
在使用bamhi和nhei消化后,vrc01、vrc01c-、3bnc60scfv在ef1α启动子的控制下被克隆入第3代慢病毒表达载体,使得scfv符合读框,其具有后接kirs2跨膜和信号传导结构域的9个氨基酸gs-连接体。dap-12位于scfv前导序列的5’,它由此处被t2a核糖体跳跃位点(ribosomalskippingsite)分隔。应用类似的策略以将scfv编码序列克隆入载体,其允许符合读框地表达scfv,其具有igg4铰链,接着是cd8跨膜结构域以及41bb共刺激和cd3ζ信号传导结构域。
基于vrc01、3bnc60、pgt128、pgdm1400和pg9scfv的cd3ζcar的构建。
使用编码5’侧翼bamhi和3’侧翼bspei位点的引物pcr扩增vrc01、3bnc60、pgt128、pgdm1400和pg9scfv编码序列,其允许消化和在前导序列和cd8α铰链之间克隆入前述的pelnscd8α铰链-cd8αtm-cd3ζ。测序验证所有构建体。
表1:序列表
慢病毒采集、浓缩、和原代cd8t细胞的转导:
质粒制剂与表达vsv糖蛋白、hiv-1gag与pol、和rev的商购的慢病毒包装质粒(prsv.rev、pmdlg/p.rre、pvsv-g)组合,并且使用lipofectamine2000转染试剂(invitrogen,lifetechnologies)以ptrpe转移载体转染至hek293t细胞上。使用rd114env和mmlvgag/pol质粒(pmscvrd114和pnglv3g/p)完成相同的过程以生成逆转录病毒。在24和48小时时间点收集上清液的30ml等分试样并且通过在4℃下以8,500rpm超速离心过夜进行浓缩。吸取上清液并且细胞沉淀物被重悬在1.2ml总体积中,在干冰上急速冰冻,并且转移至-80℃进行储存。
细胞培养–实验方案:
获得健康供体cd8和cd4t淋巴细胞并且使用磁珠——αcd3/αcd28涂覆的t-expander珠——进行活化。根据制造商的方案(stemcelltechnologies),使用rosettesep人cd4+或cd8+t细胞富集混合物通过阴性选择纯化t细胞。在补充(thermofisherscientific)有10%胎牛血清(seradigm)、1%pennstrep(lifetechnologies)、2mmglutamax(lifetechnologies)和25mmhepes缓冲液(lifetechnologies)的rpmi1640(lifetechnologies)中以1×106个/ml培养t细胞。使用3:1珠/细胞比下的抗-cd3/cd28涂覆的磁珠(lifetechnologies)和100-300国际单位/毫升的重组人白细胞介素2刺激t细胞5天并且然后去除珠。在刺激后1天,200μl的慢病毒上清液被添加至0.5×106个细胞。根据制造商的说明,在第3和5天进行mmlv载体转导,其中1ml病毒上清液添加至retronectin(takara)涂覆的24孔板并且离心感染细胞(spinoculated)。培养基在第3天加倍并且在第5天完全更换,并且然后在整个细胞培养过程中每隔一天添加,或基于细胞计数根据需要添加。
hiv-1感染。
在去除抗-cd3/cd28珠后两天,使用ccr5嗜性hiv毒株bal感染cd4t细胞,并且24小时之后以不同的效应物/靶标(e:t)比与car+cd8t细胞共培养。通过从抗cd3/cd28活化的cd4t细胞采集无细胞上清液来制备ccr5嗜性hiv-1bal病毒原液(280ng/mlp24)。在去除珠后2-3天,通过添加大约1ml的上清液/2千万个细胞来感染活化的cd4t细胞。接下来的一天,以不同的e:t比共培养cd4和cd8t细胞,并且通过细胞内p24染色监测hiv传播,根据制造商的说明,使用kc57抗-gag-rd1抗体(beckmancoulter)和invitrogenfixandperm缓冲液检测该细胞内p24染色。
体外细胞毒性和细胞因子试验
使用51cr-释放试验测试表达hiv-1gp120/41包膜(yu-2)的k562细胞的体外杀伤。5×105个靶标细胞装载有50μci的na251cro4(perkinelmer)持续90-120分钟,洗涤两次并且重悬在具有5%fbs的无酚红培养基中。转导car或模拟对照的t细胞(在初始活化后8-10天)与装载的靶标细胞以多种效应物:靶标(e:t)比共培养4小时,并且使用microβ2平板计数器(perkinelmer)测量释放入上清液的铬。在以1:1e:t比共培养5×105个转导ntd、cd4car、或scfvcar的t细胞与亲本k562、转导hla-dr*0401的k562、或转导hivyu2env的k562持续6小时后测量细胞内细胞因子产生。如先前描述的检测细胞因子产生(86),其使用大鼠抗人il-2apc(bdbiosciences),小鼠抗人mip-1βpercpcy5.5(bdbiosciences),小鼠抗人ifn-γfitc(bdbiosciences),和小鼠抗人cd107ape(bdbiosciences),连同invitrogenfixandperm缓冲液。在相同的体积中分析通过靶标细胞(没有效应细胞)的自发释放,并且通过使用5%最终浓度的sds处理靶标细胞来评估最大释放。百分比特异性裂解=[(实验释放–自发释放)/(最大释放–自发释放)]*100。以至少3个重复进行所有实验,并且其中原代人t细胞来自3个不同的供体。在400μl中共培养3×105个效应细胞和105个靶标细胞持续24h后,根据制造商的推荐,在一式两份的培养上清液中通过elisa(r&d)定量干扰素γ产生。
ccr5zfn破坏。
先前描述的ccr5特异性zfn被克隆入rna表达载体(beatty等cancerimmunologyresearch2,112-120(2014))。mmessagemmachinet7转录试剂盒(ambion,thermofisherscientific)被用于生成加帽的、体外转录的rna,其随后使用rneasyminikit(qiagen)纯化并且在1mg/ml下的无rnase水中洗脱。在电穿孔之前,使用opti-mem培养基洗涤正常供体cd8t细胞三次并且以1×108个/ml的最终浓度重悬。0.1ml中的10e6个t细胞与30μg的编码每种zfn的rna混合,使用ecm830electrosquarewaveporator(harvardapparatusbtx)在2mm杯皿(harvardapparatusbtx)中电穿孔,并且在使用αcd3/αcd28涂覆的磁珠(lifetechnologies)活化之前,在30℃下培育48小时。
为了测量通过ccr5zfn的基因组修饰效率,基因组dna由t细胞纯化并且被用于制备illumina深度测序的样品(wang等nucleicacidsres44,e30(2016))。简略地,使用巢式pcr方法以下列2种ccr5特异性引物对扩增ccr5靶标区和添加miseq接头。ccr5out-out1引物:
ctgtgcttcaaggtccttgtctgc(seqidno:80)和
ctctgtctccttctacagccaagc(seqidno:81);ccr5miseq接头引物:acacgacgctcttccgatctnnnnngccaggttgagcaggtagatg(seqidno:82)和
gacgtgtgctcttccgatctgctctactcactggtgttcatcttt(seqidno:83)。然后使用条形码引物对在后续pcr反应中添加序列条形码。对于分析基因修饰水平,自定义编写的计算机脚本被用于合并双末端150bp序列,并且经由seqprep截短接头(johnst.john,https://github.com/jstjohn/seqprep)。测序数据(read)与野生型模板序列比对。使用下列标准过滤合并的测序数据:5’和3’末端(23bp)必须精确地匹配预期的扩增子,测序数据必须不定位至靶标基因组中的不同基因座,如由bowtie2(yu等journalofvirology81,1619-1631(2007))使用默认设定测定的,并且缺失必须<70%的扩增子大小或<70bp长。如先前描述的定义比对序列中的插入缺失事件(gabriel等natbiotech29,816-823(2011)),除长度为1bp的插入缺失以外,其也被认为是真实的插入缺失以避免少算实际事件,并且真实的插入缺失必须包括横跨每个伴侣zfn的结合位点的倒数第二个碱基(邻近缺口)之间的序列内出现的缺失。
人源化小鼠模型。
6周龄nsg(nod-scidil2rgnull)小鼠获得自thejacksonlaboratory(jax)并且在7周时使用与pbs1:1混合的30mg/kg白消安进行处理。24小时之后,使用pbs中100μl0.5%人血清白蛋白中的10×106个人淋巴细胞经由尾静脉注射小鼠。三周之后,使用与pbs1:1混合的15ng的ccr5嗜性balhiv毒株经由尾静脉注射感染小鼠。通过眼眶后出血获得外周血,并且如先前描述的,使用bd裂解缓冲液和bdtrucount管枚举人cd4和car+cd8淋巴细胞计数(richardson等moleculartherapy:thejournaloftheamericansocietyofgenetherapy22,1084-1095(2014)),使用小鼠抗人cd45percpcy5.5(bdbiosciences)、小鼠抗人cd4bv421(biolegend)、和小鼠抗人cd8αbv711(biolegend)进行染色。
hiv病毒载量试验。
使用如(cillo等jclinmicrobiol52,3944-3951(2014))中描述的方法,取决于利用度从10-30μl的血浆提取rna,并且在15μl的最终体积中重构。在提取之前,均一数量的有复制能力的禽肉瘤(replicationcompetentaviansarcoma)(rcas)病毒穿刺入每个血浆样品并且单独地扩增以验证病毒/rna恢复和pcr抑制的缺乏(palmer等jclinmicrobiol41,4531-4536(2003))。使用随机六聚体逆转录rna,并且使用体外转录的rna标准品以lightcycler480probesmaster(roche;indianapolis,in)在abi7500fast实时热循环仪上通过q-pcr进行定量。对于每种样品,对5μlrna以一式两份运行q-rt-pcr反应;使用2.5μlrna对一个孔/样品运行非逆转录酶反应和rcas扩增。如(cillo等jclinmicrobiol52,3944-3951(2014))中描述的,hiv-1引物/探针靶向pol基因和检测所有m组分支,并且rcas扩增使用如(palmer等jclinmicrobiol41,4531-4536(2003))中描述的引物/探针。hiv-1定量被归一化至等价体积的初始血浆。
现在描述实验的结果。
实施例1:慢病毒骨架强化cd4car表达和对hiv复制的控制。
测试在先前的临床试验中使用的cd4car的临床前研究显示,表达此构建体的t细胞具有与自然生成的hiv特异性t细胞等价的抗病毒活性。这些临床试验展现持久性临床应答失败的假设是在这些临床试验中使用的t细胞不比内源性hiv特异性t细胞应答更强力。本发明以步进方式优化了car的每个组分以强化t细胞控制hiv复制的效力。
使用原始鼠逆转录病毒载体(mmlv)临床试验构建体和类似的第三代慢病毒载体构建体——其也使用pgk启动子以驱动cd4car表达——转导原代人cd8t细胞。与鼠逆转录病毒比较,原代人cd8t细胞的慢病毒转导一致地导致~10-倍更高的car表达的中值荧光强度(mfi)。
具有慢病毒载体、ef1α启动子、和cd8α跨膜结构域的当前新的、改进的cd4膜结合嵌合受体(cd4ζ构建体)在比具有pgk启动子和cd4跨膜结构域的构建体(在1:5和1:10下失控)低得多的e:t比下(直到大概1:100)控制hiv-1。甚至鉴于与逆转录病毒pgkcd4tmcar比较更高表达的慢病毒pgkcd4tmcar,在对hiv-1复制的控制方面可见小益处。重要地,较高表达的ef1αcd8tm构建体不促进较高的感染速率,这是因为cd8保持未感染直到大约1:100比率,而pgkcd4tm改造的cd8细胞在较低的e:t比下变得感染(图2a-2d)。在将转导cd4car的cd8t细胞稀释至较低的e:t比之后,与转导逆转录病毒载体的t细胞比较,转导慢病毒载体的t细胞在控制hiv复制上更优越(图2f-2g)。最后,转导的cd8t细胞群在1:50e:t比下均不能控制hiv传播。因而,本发明启动子和跨膜改变改进cd4膜结合嵌合受体功效和提供对hiv-1感染的增加的控制。
与转导cd4car的cd8t细胞易受无细胞病毒感染(liu等journalofvirology89,6685-6694(2015);zhen等moleculartherapy:thejournaloftheamericansocietyofgenetherapy23,1358-1367(2015))的最近报道形成对比,本结果仅显示在使用感染hiv的cd4t细胞稀释至低e:t比后检测carcd8t细胞中的细胞内gag(图2e)。相对于未转导的cd8t细胞,高水平的细胞内gag被显示存在于转导car的cd8t细胞中,其指示cd4car致使这些细胞易受感染(图2e)。重要地,较高表达的转导慢病毒的cd8t细胞上的cd4不促进这些细胞的较高感染水平或较高e:t比下的感染。此数据证明慢病毒载体是引入将t细胞重定向至靶标hiv的构建体的优选方法。
实施例2:ef1α启动子和cd8α跨膜结构域改进car表达和对hiv-1的控制。
ef1αcd8tm构建体被显示在最低e:t比下控制hiv-1复制。具有ef1α启动子(图3a-3b,中柱)的cd4tm的单纯增加的表达改进了对hiv-1复制的控制,但是ef1α启动子和cd8α跨膜置换的增加的表达的组合产生对hiv-1的最佳控制。另外,cd8αtm似乎减少cd4膜结合嵌合受体cd8细胞的感染(图3a-3b右柱集),特别是在1:50比率(ef1α构建体)和1:25比率(pgk构建体)下。
cd8αtm结构域的置换降低car+cd8t细胞的感染,而不管在1:25和1:50e:t比下使用的启动子(图3b)。然而,如图2a-2g中可见,car+cd8t细胞可以被稀释至它们不再可以控制hiv感染和使其屈服于感染的点。因而,相对于临床试验逆转录病毒,改变病毒载体、启动子、和跨膜结构域给予50-倍效力增加,其导致在1:50e:t比下对hiv复制的体外完全控制(图3c-3d)。
因而,cd8α跨膜改变是改进hiv-1复制控制的至关重要的修饰。
实施例3:基于广泛中和抗体的car可以控制hiv-1
如图4a-4b中可见,基于单链可变片段(scfv)广泛中和抗体(bnab)的car和cd4膜结合嵌合受体二者可以控制hiv-1感染。以它们的hiv-1结合宽度和中和效力排列的一组scfv(参见图4a中的表格)被克隆入相同的ef1α-cd8αtm-ζ构建体骨架作为cd4膜结合嵌合受体,并且评估它们控制hiv-1复制的能力。来自4小时共培养的cr51释放杀伤试验显示了表达scfvcar或cd4膜结合嵌合受体中任一种的cd8t细胞,而非未转导的(ntd)t细胞、裂解的表达env的靶标细胞。这指示scfvcar以适当的折叠/构象表达并且能够结合env(图4b)。scfvcar中的许多响应于表达env的靶标一致地产生高水平的细胞内细胞因子。cd4膜结合嵌合受体和scfvcar二者展现了相对于ntd对照高至1:50-1:200的e:t比的env靶标细胞的裂解(图4c),其优于在图1中显示的kf11tcr的活性,该kf11tcr展现了高至1:25e:t比的功效。
不希望受限于理论,理解使用靶向hiv的car,其中car包括抗-hivscfv将有益于治疗具有感染hiv的细胞的储存群体的对象。本发明因此在本发明的一方面包括用于这样的用途的这样的car。
实施例4:cd4car在体外比hiv特异性精英控制者tcr强力超过100-倍。
精英控制者(ec)是在不存在art的情况下能够控制hiv复制的罕见个体。某些hla等位基因,比如hla-b57,在精英控制者组群中超出比例,这表明t细胞应答在控制这些个体中的hiv复制中发挥关键作用(walker等naturereviews.immunology13,487-498(2013))。由ec分离的hiv特异性t细胞比由hiv进展者分离的hiv特异性t细胞具有更高的细胞溶解潜力(migueles等pnas97,2709-2714(2000);saez-cirion等pnas104,6776-6781(2007))。因此,为了确定表达cd4car的t细胞是否能够比由精英控制者——其与患者中对hiv复制较好的控制相关联——分离的表达hla-b57局限的tcr的t细胞更好地控制hiv复制,表达hla-b57的原代人cd8t细胞转导特异于kf11(hivp24gag表位kafspevipmf,seqidno:42)的tcr——其也在ef1α启动子下表达,并且使用来自相同供体的cd4t细胞测定它们限制hiv传播的能力。kf11tcr在本领域已知与患者中对hiv复制较好的控制相关联并且是针对hiv-1的最强力的患者源tcr之一。虽然转导kf11tcr的cd8t细胞低至1:25e:t比降低hiv复制,但是从没有取得对hiv复制的完全控制(图1a-1b和图3a-3d)。相比之下,本发明的cd4car低至1:100e:t比几乎完全控制hiv。这些结果揭示了cd4ζcar比hla-b*57局限的kf11精英控制者tcr更好地控制nl4-3hiv-1复制。
这些数据指示本发明的cd4car比内源性hivt细胞应答具有大得多的效力并且表明表达优化的cd4car的t细胞在art去除后在通过过继性t细胞疗法能够取得的效应物/靶标比下将能够控制hiv复制。
另外,图5a-5b揭示了cd28共刺激促进对hiv-1bal的控制。与cd4ζ构建体同样地或更好地一致控制hiv-1的另一种膜结合嵌合受体构建体是cd4cd28ζ构建体。
实施例5:cd28和4-1bb共刺激对体外hiv-1复制的控制具有相反作用。
t细胞需要增殖、效应物功能、和长期存活的共刺激信号。另外的共刺激结构域,比如cd28和4-1bb,已经被并入最近的car设计,其已经证明对体外诱导持久性cart细胞应答是必需的(vanderstegen等naturereviews.drugdiscovery14,499-509(2015))。生成包含各种共刺激结构域的一组cd4car,连同cd3-ζ结构域,包括cd28、4-1bb、cd28+4-1bb、ox40、icos、或cd27,并且测试它们体外控制hiv感染的能力。表达car——其包含4-1bb、cd27、或icos共刺激结构域——的cd8t细胞不如表达car——其仅包含cd3-ζ信号传导结构域——的t细胞一样有效地控制hiv,这表明这些共刺激途径干扰hiv复制的t细胞控制(图5a-5b)。包含ox40或cd28和4-1bb的组合的car以与仅包含cd3-ζ的car的类似水平控制hiv。相比之下,cd28改进体外控制,其指示cd28共刺激在hiv特异性car中可能是有利的。
实施例5:cd4car特异性地应答于env+细胞而非ii类mhc+细胞。
临床前数据展现了表达临床试验cd4car的t细胞不杀伤raji细胞,其表达高水平的ii类mhc——cd4的低亲和力配体(romeo等cell64,1037-1046(1991))。然而,患者中cd4cart细胞的长半衰期被推测是由于与ii类mhc的低亲和力相互作用或可能由于在病毒信号(viralblip)期间释放env(scholler等,sciencetranslationalmedicine4,132ra153(2012))。为了确定先前描述的载体修饰是否有助于对ii类mhc的脱靶反应性,针对稳定表达高水平的hla-dr*0401等位基因的k562细胞系测量cd4car+cd8t细胞应答(参见图14)。cd8t细胞转导优化的cd4car——其包含cd3-ζ、4-1bb-cd3-ζ、或cd28-cd3-ζ共刺激结构域——并且与未修饰的k562靶标细胞、hla-dr*0401+k562细胞、或hivyu2env+k562细胞一起培养。转导car的cd8t细胞响应于hivenv+靶标而不响应于hla-dr+或亲本k562产生il-2、cd107a、ifn-γ、和mip-1β,其中最稳健的产生在包含cd28的cart细胞中(图11a)。当与亲本或表达hla-dr的靶标混合时,对于所有car观察到少量的mip-1β信号,其在未转导的对照中没有观察到,这可能是由于在表达car的t细胞中观察到的一些组成型、非抗原特异性信号传导。然而,与单独或与亲本k562一起培养的cart细胞比较,响应于表达ii类mhc的细胞没有检测到另外的细胞因子或cd107a产生。
进行共培养试验以确定car+cd8t细胞是否可以杀伤ii类mhc靶标,而不管缺乏细胞因子产生。包含cd28-cd3-ζ共刺激结构域的cd4car被添加至hla-a*02+/gfp+k562和hla-dr*0401+/mcherry+k562的1:1混合物,并且随着时间测量两种k562细胞类型的比率。72小时内延长的培养不导致hla-dr*0401+k562的比例减小(图11b-11c)。这些结果表明使用再改造的cd4car将和原始cd4car载体一样安全地用于人。
实施例6:与第一代cd4car相比,表达优化的cd4car的t细胞在体内控制hiv-1复制并且扩展至大得多的水平。
与包含4-1bb和cd3-ζ信号传导结构域的那些相比,包含cd28和cd3-ζ信号传导结构域的cd19特异性car具有更优越的体外活性,但是包含4-1bb的car证明在人源化小鼠模型和最终在患有b细胞白血病的患者中更优越((porter等sciencetranslationalmedicine7,303ra139(2015);milone等moleculartherapy:thejournaloftheamericansocietyofgenetherapy17,1453-1464(2009);brentjens等blood118,4817-4828(2011))。为了确定这对靶向hiv的car是否符合,以及本发明的优化的cd4car是否可以比原始cd4car构建体——其先前在临床中测试——更好地体内控制hiv,小鼠的组群输注有1千万个人t细胞,包括8百万个cd4t细胞和2百万个cd8t细胞。四组小鼠与不同的cd8效应细胞群比较:未转导的(ntd),转导包含cd28(28z)或4-1bb共刺激结构域(bbz)的最佳慢病毒载体,或转导临床试验mmlv基载体(cd4z)。
感染前基线cd4t细胞计数在ntd、bbz、或28z组之间不显著不同,并且cd4z处理的小鼠显著更高(图12a)。然后以ccr5嗜性hiv毒株bal感染小鼠,并且在22天的感染后枚举终点外周cd4t细胞计数。输注表达bbz或28z构建体的t细胞的小鼠分别显示了人cd4t细胞数目的17-倍和177-倍扩展(图12b)。相比之下,外周cd4计数在使用未转导的cd8t细胞或cd4zt细胞处理的小鼠中保持非常低,这大概是由于hiv介导的耗尽(图12b)。不同小鼠组群中cd4car+cd8t细胞数目的检查分别揭示了bbz、28z、和cd4zt细胞的389-倍,587-倍、和2-倍扩展(图12c-12d)。还在此模型中检查cd4cart细胞控制病毒载量的能力。在hiv感染之后七天,bbzcart细胞展示了对病毒复制的最大控制,以及最不可检测到的病毒载量,而来自ntd、28z、和cd4z处理的动物的血浆包含大约1个拷贝的hivrna/μl(图12e)。感染之后18天,与使用ntdt细胞处理的小鼠比较,hivrna的中值拷贝数在bbz和28z处理的动物中降低超过10倍(图12f)。然而,与ntd处理的小鼠比较,cd4z处理的小鼠不在统计学上减少hivrna。虽然任一种优化的cd4car比临床试验car更好地控制hiv,但是这些结果指示不同的共刺激结构域在hiv感染中具有各异的保护作用,其中cd28以最大程度保护cd4t细胞和4-1bb促进对hiv复制的早期控制。
实施例7:体内富集ccr5-zfn修饰的cd4carcd8t细胞。
不管对hiv复制增加的体外和体内控制,体外数据表明使cd8t细胞转导hiv特异性car致使它们易受感染。为了确定这些cart细胞是否在体内变得感染,在存在hiv的情况下体内测试cart细胞抵抗感染富集。先前已经在原代人cd4t细胞中证明了通过使用锌指核酸酶(zfn)基因编辑ccr5hiv共受体给予的存活益处(perez等natbiotechnol26,808-816(2008);tebas等naturereviews.immunology13,693-701(2013))。然而,还没有探究在存在hiv感染的情况下对cd4car+cd8t细胞的类似的富集分析。为了确定共受体编辑是否促进更好的car+cd8t细胞富集,作为确定cd4carcd8t细胞在体内易受感染的手段,四个额外的组被包括在图12a-12f中描述的实验中,其中cd8t细胞使用ccr5zfn进行处理并且随后转导cd4car构建体中的每种。
相对于未转导的cd8t细胞,在使用cd4cart细胞处理和感染hiv的小鼠中富集ccr5破坏的等位基因(图13a)。植入有未转导的cd8t细胞的感染hiv的小鼠具有0.4%的中值破坏频率。由于这些cd8t细胞不是转导cd4car的,它们不预期在存在hiv的情况下富集。使用bbz、28z、或cd4zt细胞处理的小鼠分别具有14.4%、29.6%、和9.5%的中值破坏频率,其指示hiv-抗性、表达cd4car的cd8t细胞在存在hiv感染的情况下富集。ccr5破坏富集受car+cd8t细胞在存在hiv的情况下扩展的能力影响,如图13b和图15中显示的,并且因而28zcart细胞的较大的抗原特异性扩展可以负责图8a中可见的较大的ccr5富集。此数据还显示bbz改造的t细胞在不存在抗原的情况下能够比28z改造的t细胞更好地持续,其与使用肿瘤特异性car的发现一致(milone等moleculartherapy:thejournaloftheamericansocietyofgenetherapy17,1453-1464(2009);carpenito等pnas106,3360-3365(2009))。总之,此数据表明发生hiv特异性cart细胞的hiv感染,并且防护这些细胞免于hiv感染的策略将可能对确保hiv感染的持久性控制是必需的。
还检查了致使cd4carcd8t细胞抵抗hiv感染是否增强它们控制hiv-1感染的能力。在22天的感染后,一般而言,在感染的小鼠给出的zfn处理的细胞或非zfn处理的细胞中,在外周血cd4t细胞计数或car+cd8t细胞的扩展中不存在显著差异,除与非zfn处理的比较使用zfn处理的28z小鼠中更低的终点cd4t细胞计数以外(图16a-16b)。然而,在zfn处理的cd4z小鼠中存在较大的cd4和car+cd8t细胞扩展的趋势(图16a-16b)。在28z或bbz小鼠中,其在不存在zfn处理的情况下具有低病毒载量,没有zfn处理造成的病毒rna的进一步减少(图13c)。另一方面,在给予zfn处理的cd4zt细胞的小鼠中可见血浆病毒rna的显著减少,到了血浆病毒不再显著高于给予bbz或28zcar的小鼠的地步。由于与不具有zfn的bbz或28z处理的小鼠比较,病毒载量在不具有zfn的cd4z逆转录病毒处理的小鼠中是大约log-倍更高,在由zfn处理造成的差控制的病毒复制的背景下,较容易检测到病毒载量的显著降低。而且,短的实验持续时间可以已经排除可见zfn处理对cart细胞的完整益处,包括可检测的转导的cd8t细胞的富集,特别是在cd4z处理的小鼠中,其中它们产生最大抗病毒益处。总之,在不存在zfn处理的情况下,优化的car在体内优于临床试验cd4z;然而,如果表达原始car构建体的t细胞可以由ccr5破坏保护,则可以在此相对短的体内试验内取得对hiv复制的控制。
总之,本文公开的改进的hiv复制控制是印象深刻的(图17a-17c)。本发明包括对先前公开的cd4膜结合嵌合受体的数种改进,其导致杀伤感染hiv-1的细胞的能力的50-倍增加。此增加的功效,与第1代cd4膜结合嵌合受体不同,滴定至在体外试验中生理学相关的效应物:靶标(e:t)比,这表明这些改进的cd4膜结合嵌合受体构建体将可能在降低患者中的病毒载量中更有效。
长期控制hiv感染的主要挑战之一是一些病毒突变体的免疫逃逸。本发明的基于scfv的car(scfv-car)解决此挑战并且被显示有利于治疗具有感染hiv的细胞的储存群体的对象。添加表达基于scfv的car的t细胞可以进一步强化改造的t细胞在不存在art的情况下提供持久性hiv复制控制的能力。如图9a和图10a中可见,vrc01、vrc01c和3bnc60-car显示了表达hiv-1包膜gp120/41(yu-2分离株)的k562细胞而非表达cd19的k562细胞的特异性裂解,其确认scfv-car细胞毒性是强力的和特异性的二者。而且,由scfv-cart细胞释放的干扰素γ(ifng)与细胞的杀伤功效紧密关联(图9c和图10c)。另外,通过使用杀伤细胞免疫球蛋白样受体(kir)细胞质结构域进一步增加scfv-car细胞毒性。
本发明提供了改进的hiv疗法的众多潜力。由hiv患者分离的t细胞可以转导scfv-car和/或cd4膜结合嵌合受体,离体扩展和再输注入对象。转导的t细胞识别表达gp120的细胞并且成为活化的,其导致杀伤带有hiv的细胞。此途径可以被应用于任何病毒介导的疾病,其中靶标抗原可以转移或漂移,并且因此逃逸通过正常抗体(例如甲型流感的血凝素)的免疫监督。此疗法可以与其它已知的抗体疗法haart组合,或可以在car疗法期间或之前与病毒诱导物组合。
本发明的预防性应用在高风险群体中也是可能的,由于t细胞应当杀伤感染的cd4细胞,其防止建立hiv所有组成成分。
其它实施方式
本文对变量的任何定义中要素列表的叙述包括该变量作为任何单个要素或列出的要素的组合(或子组合)的定义。本文对实施方式的叙述包括作为任何单个实施方式或与任何其它实施方式或其部分组合的该实施方式。
本文引用的每个和每种专利、专利申请、和出版物的公开内容由此通过引用以其全部并入本文。虽然已经参照具体的实施方式公开了本发明,但是明显的是,本领域技术人员可以设计本发明的其它实施方式和变型而不背离本发明的真实精神和范围。所附权利要求意图解释为包括所有这样的实施方式和等价变型。
- 还没有人留言评论。精彩留言会获得点赞!