一种间三氟甲基苯基丙醇的制备方法与流程
本发明涉及一种间三氟甲基苯基丙醇的制备方法,属于医药化工技术领域。
背景技术:
间三氟甲基苯基丙醇是药物西那卡塞的中间体,西那卡塞是被称为拟钙剂的新一类化合物中第一个药物,能够激活甲状旁腺中的钙受体,从而降低甲状旁腺素的分泌。西那卡塞的结构式为A,它由两个片段组合而成,一部分为R-1-萘基乙胺,另一部分为间三氟甲基苯基丙基类化合物,合成A有多种方法,代表性路线有四种,如图1所示:
路线1:间三氟甲基苯基丙酸与R-1-萘基乙胺形成酰胺,再还原酰胺键得A(MaceleodsPharma.,WO 2008/117299;Dr.Reddy’s Lab.,WO 2008/58235);
路线2:间三氟甲基苯基丙醛与R-1-萘基乙胺形成席夫碱,再还原C-N双键得A(Medichem,S.A.,WO 2008/68625;Dipharma,EP2327684);
路线3:间三氟甲基苯基丙醇与酰氯形成活化酯,再与R-1-萘基乙胺进行取代反应得A(Cipla,WO2010/100429;Teva,WO2008/63645);
路线4:间三氟甲基肉桂酸与R-1-萘基乙胺形成肉桂酰胺,再还原得到A(Amgen,WO2009/2427)。
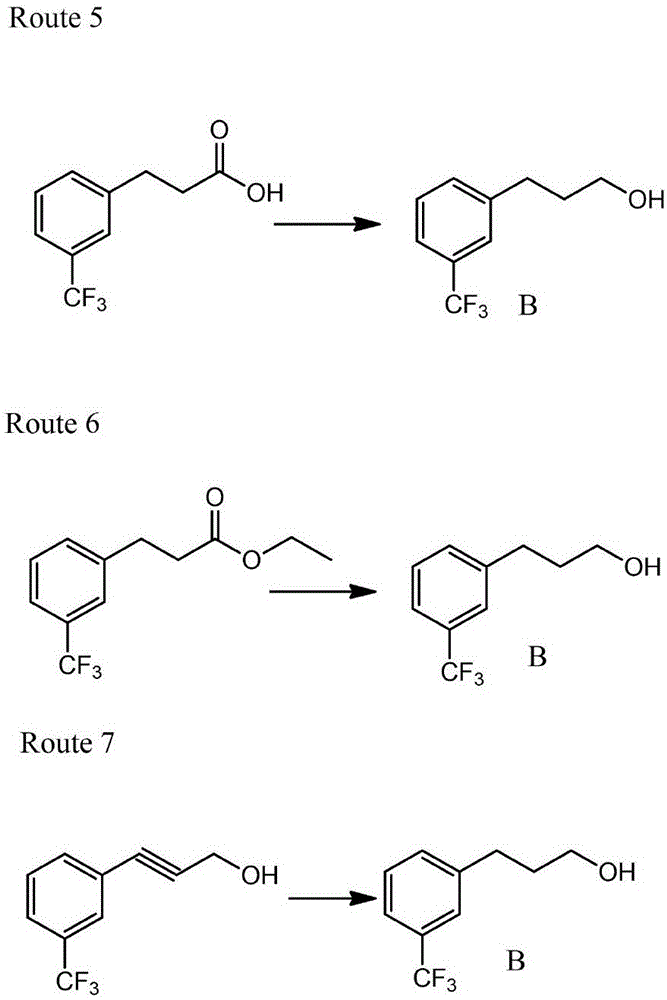
而针对于间三氟甲基苯基丙醇的合成,即结构为B,各类文献也多有很多报道,具有代表性的合成方式有三种,如图2所示:
路线5:从间三氟苯基丙酸出发,用BH3/THF还原得到B,(ActelionPharma.,US2009/99228);用BH3/Me2S还原得到B,(Cipla,WO2010/100429);用LiAlH4还原得到B,(Dr.Reddy’s Lab.,WO2008/58235);
路线6:从间三氟甲基苯基丙酸乙酯出发,用NaBH4还原得到B,(Teva,WO2006/125026);
路线7:从间三氟甲基苯基丙炔醇出发,用Pd/C加氢还原得到B,(Medichem,S.A.,US2010/267988)。
如何通过工业化生产高效、简单获取间三氟甲基苯基丙醇是亟待解决的问题。
技术实现要素:
本发明提供一种间三氟甲基苯基丙醇的制备方法,解决现有技术中间三氟甲基苯基丙醇制备操作复杂、耗用还原剂量大的技术问题。
为了实现本发明的目的,技术方案具体包括如下步骤:
(1)将间三氟甲基苯基丙酸和有机胺加入有机溶剂中,温度控制在-10℃~30℃;
(2)在步骤(1)获得溶液中滴加卤代甲酸酯,温度控制在-10℃~-5℃,卤代甲酸酯滴加时间为15min~20min,反应时间为30min,过滤去除有机胺盐酸盐,获得活化混合酸酐溶液;
(3)在步骤(2)获得的混合酸酐溶液中滴加碱金属硼化物的还原剂溶液,温度控制在0℃~5℃,碱金属硼化物的还原剂溶液滴加时间为1.5h~2h,还原反应时间为1h,还原反应结束后,减压浓缩,加二氯甲烷萃取两次,合并有机相,用1当量浓度的NaOH溶液洗涤两次,饱和食盐水洗两次,有机相用无水硫酸钠干燥1h~10h,有机相过滤后浓缩二氯甲烷,浓缩液用2mmHg减压蒸馏获得无色透明液体间三氟甲基苯基丙醇。
本发明中,步骤(1)中所述有机胺为三级脂肪胺、三级芳香胺中一种或多种,所述三级脂肪胺为三乙胺、二异丙基乙胺、苄基二甲基胺、N-甲基吗啡啉中一种或多种,所述三级芳香胺为吡啶、N,N-二甲基-4-胺基吡啶中一种或多种,优选三乙胺和N-甲基吗啡啉,所述间三氟甲基苯基丙酸与所述有机胺的物质的量比例为1.05~1.2倍,优选1.1倍。
本发明中,步骤(1)中所述有机溶剂为卤代烃溶剂、醚类溶剂中一种或多种,所述卤代烃溶剂为二氯甲烷、三氯甲烷、1,2-二氯乙烷中一种或多种,所述醚类溶剂乙醚、异丙醚、二甘醇二甲醚、二氧六环、四氢呋喃、2-甲基四氢呋喃中一种或多种,优选2-甲基四氢呋喃、二甘醇二甲醚,特别优选2-甲基四氢呋喃,所述有机溶剂中的反应物浓度为0.2mol/L~0.8mol/L,优选0.5mol/L。
本发明中,步骤(2)中所述卤代甲酸酯为氯甲酸甲酯、氯甲酸乙酯、氯甲酸异丙酯、氯甲酸异丁酯中一种或多种,优选氯甲酸乙酯、氯甲酸异丁酯,特别优选氯甲酸异丁酯,所述间三氟甲基苯基丙酸与所述卤代甲酸酯的物质的量比例为1.05~1.2倍,优选1.1倍。
本发明中,步骤(3)中所述碱金属硼化物为硼氢化钾、硼氢化钠中一种或多种,优选硼氢化钠,所述间三氟甲基苯基丙酸与所述碱金属硼化物的物质的量比例为1~2.5倍,优选1.5倍。
本发明中,步骤(3)中所述还原剂溶液为所述碱金属硼化物的水溶液、所述碱金属硼化物与四氢呋喃溶液、所述碱金属硼化物和四氢呋喃及水的混合溶液、所述碱金属硼化物与二甘醇二甲醚的悬乳液中一种或多种,优选所述碱金属硼化物的水溶液。
本发明的有益效果:
1、在羧酸形成混合酸酐后,采用碱金属硼化物还原到醇,相对现有技术而言,更简单、易于操作,而且耗用的还原剂量更小,更为经济;
2、本发明制备方法具有原料价廉易得、方法简便、收率能达到92.2%,纯度达到99.8%,且酸酐还原成醇的选择性高,适合工业化生产;
3、本发明适合的温度在-10℃~30℃范围内,反应条件温和,在步骤(2)中酸酐形成过程中温度控制-10℃~-5℃,在步骤(3)中酸酐还原成醇过程中温度控制在0℃~5℃,温度控制容易实现;
4、本发明操作简单,不涉及高温高压反应,一般常规性化工生产设备就能完成生产,对设备要求低,适宜批量化生产;
5、萃取、干燥、洗涤等采用常规性溶剂就能实现,如二氯甲烷、氢氧化钠溶液、饱和食盐水、无水硫酸钠等,具有毒性小,来源方便,价格低廉,容易回收等特点。
附图说明
图1示出了现有技术结构A合成路线。
图2示出了现有技术结构B合成路线。
具体实施方式
以下将参照实例对本发明作进一步的详细描述,但本发明不限于这些具体实例。
本发明的目标产物为间三氟甲基苯基丙醇,其纯度用高压液相色谱进行检测。
具体检测条件为:
流动相A:含有0.05%三氟乙酸的水,流动相B:色谱级乙腈
配样:样品25mg溶解在25mL1:1的A与B的混合溶液中;
检测柱:Zorbax Eclipse XDB C18,150mm×4.6mm,5μm;
柱温:室温;
检测波长:220nm;
流动相:0-8min,B,30%;8.1-13min,B,90%;13.1-15min,B,30%;
流速:1.0mL/min;
进样量:5μl;
运行时间:15min;
样品的保留时间为8.41min,原料的保留时间为5.86min;
纯度以面积归一法确定。
具体实施例1:
250mL三口烧瓶中,加入10.9克间三氟甲基苯基丙酸(50mmol),100mL 2-甲基四氢呋喃,5.6克N-甲基吗啡啉(55mmol,1.1eq.),冷至-10℃,滴加5.9克氯甲酸乙酯(55mmol,1.1eq.),控制温度在-5℃以下,约20min加完,反应30min,过滤除去生成的N-甲基吗啡啉盐酸盐,滤液中滴加2.85克NaBH4(75mmol,1.5eq.)/20克水的溶液,约2h加完,控制温度在0-5℃,反应1h完毕。反应结束后,减压浓缩去溶剂,加100mL二氯甲烷萃取两次,合并有机相,1N NaOH 50mL洗两次,饱和食盐水50mL洗两次,有机相用5克无水硫酸钠干燥1h,过滤后浓缩二氯甲烷,残液2mmHg减压蒸馏得无色透明液体8.7克,收率85.3%,纯度99.7%。
1H-NMR(300MHz,CDCl3),ppm:7.45-7.31(m,4H),3.69(t,2H,J=6.3Hz),2.77(t,2H,J=7.7Hz),1.90(m,2H)。
实施例2:
250mL三口烧瓶中,加入10.9克间三氟甲基苯基丙酸(50mmol),100mL二氯甲烷,5.6克N-甲基吗啡啉(55mmol,1.1eq.),冷至-10℃,滴加5.9克氯甲酸乙酯(55mmol,1.1eq.),控制温度在-5℃以下,约20min加完,反应30min,过滤除去生成的N-甲基吗啡啉盐酸盐,滤液中滴加2.85克NaBH4(75mmol,1.5eq.)/20克水的溶液,约2h加完,控制温度在0-5℃,反应2h完毕。反应结束后,1N NaOH50mL洗两次,饱和食盐水50mL洗两次,有机相用5克无水硫酸钠干燥1h,过滤后浓缩二氯甲烷,残液2mmHg减压蒸馏得无色透明液体7.8克,收率76.5%,纯度99.6%。
实施例3:
250mL三口烧瓶中,加入10.9克间三氟甲基苯基丙酸(50mmol),100mL二甘醇二甲醚,5.6克三乙胺(55mmol,1.1eq.),冷至-10℃,滴加7.45克氯甲酸异丁酯(55mmol,1.1eq.),控制温度在-5℃以下,约20min加完,反应30min,过滤除去生成的三乙胺盐酸盐,滤液中滴加3.8克NaBH4(100mmol,2eq.)/20克二甘醇二甲醚的悬浮液,约2h加完,控制温度在0-5℃,反应3h完毕。反应结束后,减压浓缩去溶剂,加100mL二氯甲烷萃取两次,合并有机相,1N NaOH 50mL洗两次,饱和食盐水50mL洗两次,有机相用5克无水硫酸钠干燥2h,过滤后浓缩二氯甲烷,残液2mmHg减压蒸馏得无色透明液体9.3克,收率91.2%,纯度99.8%。
实施例4:
250mL三口烧瓶中,加入10.9克间三氟甲基苯基丙酸(50mmol),100mL二甘醇二甲醚,5.6克N-甲基吗啡啉(55mmol,1.1eq.),冷至-10℃,滴加5.9克氯甲酸乙酯(55mmol,1.1eq.),控制温度在-5℃以下,约20min加完,反应30min,过滤除去生成的N-甲基吗啡啉盐酸盐,滤液中滴加4克KBH4(75mmol,1.5eq.)/20克二甘醇二甲醚的悬浮液,约2h加完,控制温度在0-5℃,反应4h完毕。反应结束后,减压浓缩去溶剂,加100mL二氯甲烷萃取两次,合并有机相,1N NaOH 50mL洗两次,饱和食盐水50mL洗两次,有机相用5克无水硫酸钠干燥1h,过滤后浓缩二氯甲烷,残液2mmHg减压蒸馏得无色透明液体8.6克,收率84.2%,纯度99.7%。
实施例5:
2000mL三口烧瓶中,加入109克间三氟甲基苯基丙酸(500mmol),900mL 2-甲基四氢呋喃,56克N-甲基吗啡啉(550mmol,1.1eq.),冷至-10℃,滴加59克氯甲酸乙酯(550mmol,1.1eq.),控制温度在-5℃以下,约40min加完,反应60min,过滤除去生成的N-甲基吗啡啉盐酸盐,滤液中滴加28.5克NaBH4(750mmol,1.5eq.)/200克水的溶液,约2h加完,控制温度在0-5℃,反应3h完毕。反应结束后,减压浓缩去溶剂,加600mL二氯甲烷萃取两次,合并有机相,1N NaOH 300mL洗两次,饱和食盐水200mL洗两次,有机相用40克无水硫酸钠干燥4h,过滤后浓缩二氯甲烷,残液2mmHg减压蒸馏得无色透明液体94.1克,收率92.2%,纯度99.7%。
- 还没有人留言评论。精彩留言会获得点赞!