一种4‑溴‑2‑氟苯胺的选择性合成方法与流程
本发明涉及一种4-溴-2-氟苯胺的选择性合成方法,具体地说是一种用就地生成的过溴型季铵盐溴化邻氟苯胺生产4-溴-2-氟苯胺的方法,属于精细化工及药物中间体合成领域。
背景技术:
4-溴-2-氟苯胺是合成镇痛消炎药氟比洛芬(Flurbiprofen)的中间体[郑庚修,嵇耀武,黄志新.氟比洛芬的合成,中国医药工业杂志,1991,22(1):2-3;蔡汉兴,朱明生,邱春明,等.2-(3-氟-4-苯基)苯丙酸合成,江西化工,2006,(3):83-86]。该中间体主要通过邻氟苯胺溴化制得,但由于邻氟苯胺中的-NH2的强给电子效应,致使苯环的活性非常高,用液溴溴代易发生多溴代;另外除氨基的对位发生溴代外,其邻位也可以发生溴代。以上原因致使溴代反应的选择性很差。因此,一般采用N-溴代琥珀酰亚胺[王超莉,谭应龙,徐正安,等.4-溴-2-氟联苯合成工艺研究,江西师范大学学报(自然科学版),2007,31(2):149-152]、1,3-二溴-5,5-二甲基海因[王尊元,马臻,梁美好,等.氟比洛芬的新法合成,齐鲁药事,2005,24(11):687-688]等选择性溴代试剂进行溴代。尽管采用这些溴代试剂进行溴代产品的选择性和产率较高,但它们价格贵,溴代的原子利用率低,因而4-溴-2-氟苯胺的合成成本高,三废排放量大。
技术实现要素:
针对4-溴-2-氟苯胺合成工艺存在的问题,本发明的发明者对邻氟苯胺的选择性溴代进行了深入研究,发现利用液溴和季铵盐就地生成的过溴型季铵盐溴代邻氟苯胺生成4-溴-2-氟苯胺具有很好地选择性,且季铵盐的用量小,因而溴代成本低,三废量少。另外,溴代温度低,溴代速度快,产率高。
本发明的技术方案为:
(1)合成原理
(2)工艺步骤
1)合成:将季铵盐、邻氟苯胺和溶剂加入反应瓶中,搅拌溶解,在控制温度低于30℃的条件下,缓慢滴加溴素,约30~60min滴完,之后将物料温度升至40℃左右反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用溶剂洗涤两次,随后将滤饼分散于溶剂中,再加10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用去离子水洗涤两次后蒸出溶剂得4-溴-2-氟苯胺。
所述的邻氟苯胺和溴素的摩尔比为1:1.0~1.1,优选为1:1.4~1.7;
所述的季铵盐为四烷基溴化铵和四烷基氯化铵,如四丁基溴化铵、四乙基溴化铵、四丁基氯化铵、四乙基氯化铵等,优选为四丁基溴化铵;
所述的邻氟苯胺与季铵盐的摩尔比0.10~0.15:1;
所述的溶剂为二氯乙烷、二氯甲烷、三氯甲烷、四氯化碳、环己烷、正己烷等,优选为二氯甲烷;
所述的反应过程中所用溶剂与邻氟苯胺的质量比为2~4:1,优选为2.5~3.5:1。
所述的滤饼溶解所用溶剂与滤饼的质量比为1.0~4.0:1,优选为1.5~2.5:1。
附图说明
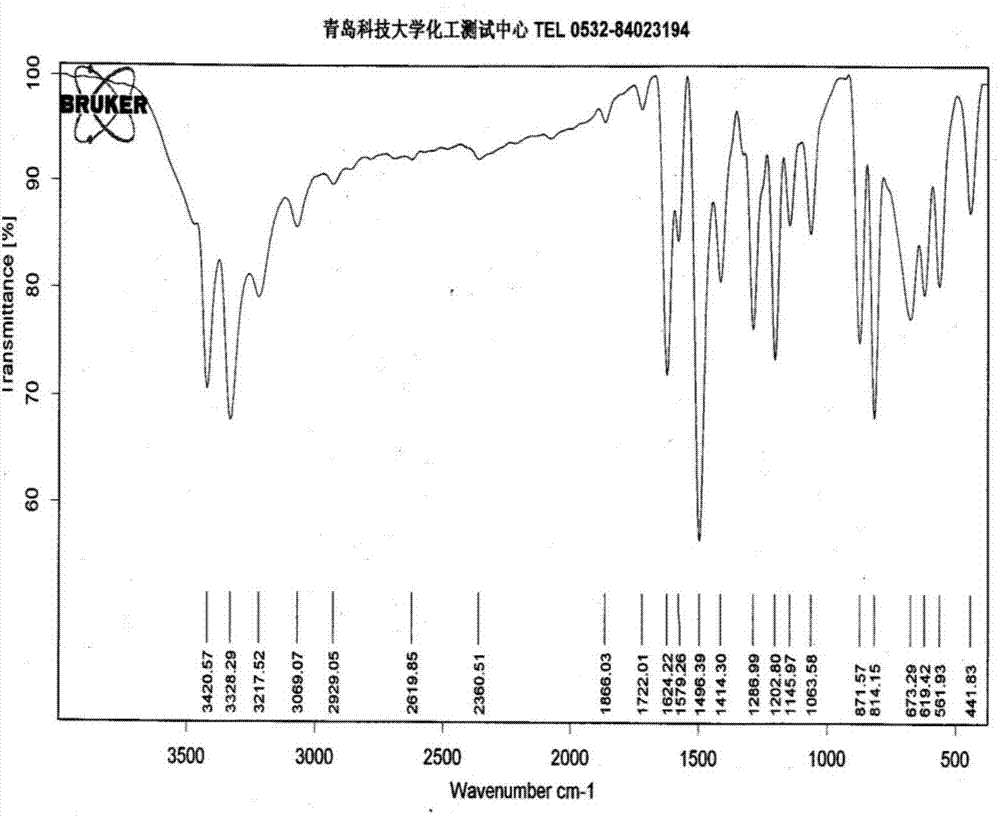
图1为本发明实施例1得到的4-溴-2-氟苯胺的红外光谱图;
图2为本发明实施例1得到的4-溴-2-氟苯胺的核磁共振氢谱图;
图3为本发明实施例1得到的4-溴-2-氟苯胺的核磁共振碳谱图;
图4为本发明实施例1得到的4-溴-2-氟苯胺的质谱图。
具体实施方式
以下对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
除非另有说明,本发明中所采用的百分数均为质量百分数。
产品含量采用气相色谱分析。气相色谱分析条件如下:柱型:AC1.10;进样温度:250℃;检测温度:280℃;流动相:氯仿;采用程序升温,每分钟10℃;数据采用浙大智能N2000数据工作站处理,含量采用面积归一法计算。
产品的核磁共振氢谱和碳谱采用德国布鲁克公司的AVANCE-500型核磁共振仪测定,均以氘代氯仿为溶剂。
红外光谱采用德国布鲁克公司的TENSOR-27红外光谱仪测定,采用溴化钾压片法测定。
质谱采用美国安捷伦科技有限公司的Agilent 19091S-433型气质联用仪测定。
实施例1
一种4-溴-2-氟苯胺的选择性合成方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入7.1g(0.022mol)四丁基溴化铵、22.2g(0.20mol)邻氟苯胺和50mL二氯甲烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至回流(约40℃)反应约2.5h。
2)后处理:冷却至室温,过滤,滤饼用20×2mL的二氯甲烷洗涤两次,随后将滤饼分散于50mL二氯甲烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得34.3g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为94.8%,产率为85.6%。
本发明还通过红外光谱、核磁共振谱和质谱测定对本实施例得到的产物结构进行了表征。图1为本发明实施例1得到的产物的红外光谱图(σ,cm-1);图2为本发明实施例1得到的产物的核磁共振氢谱图(1H-NMR,500MHz,CDCl3);图3为本发明实施例1得到的产物的核磁共振碳谱图(13C-NMR,500MHz,CDCl3);图4为本发明实施例得到的产物的质谱。
图1中,3068cm-1附近的中等强度的吸收峰为苯环上的的伸缩振动吸收峰;1624cm-1、1579cm-1、1497cm-1、1414cm-1处的四个强度不一的吸收峰为苯环骨架的振动吸收峰,这些峰的存在说明有的存在。814cm-1和872cm-1处的强吸收峰是苯环结构中1,2,4-三取代的特征峰。3420cm-1处中等强度的吸收峰为N-H伸缩振动特征峰,650~900cm-1处的吸收峰为N-H的面外弯曲振动吸收峰、1263cm-1处的吸收峰为苯环上C-N的伸缩振动特征峰,说明苯环上有-NH2的存在。以上红外光谱与目标产物的结构特征吻合。
图2中,处于δ=7.12ppm及7.03ppm的两组双重峰和δ=6.63ppm的三重峰是苯环上邻对位三取代氢的特征峰,其强度比为1:1:1,代表3个氢核,即分子式中含处于δ=3.71ppm的单峰是-NH2中氢的特征峰。以上氢谱与目标产物的结构特征吻合。
图3中,152.3ppm、133.8ppm、127.4ppm、118.5ppm、117.7ppm和108.8ppm分别是2号位、1号位、5号位、3号位、6号位和4号位的碳原子峰,与目标产物的结构特征吻合。
图4中,m/z=189是的分子离子峰。m/z=80说明有碎片存在,m/z=83说明有碎片存在,m/z=94说明有碎片存在,m/z=110说明有碎片存在,m/z=160说明有碎片存在。以上分子离子峰及碎片与目标产物的结构特征吻合。
以上综合分析表明,所得产品即为4-溴-2-氟苯胺。
实施例2
一种4-溴-2-氟苯胺的选择性合成方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入4.2g(0.022mol)四乙基溴化铵、22.2g(0.20mol)邻氟苯胺和50mL二氯甲烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至回流(约40℃)反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用20×2mL的二氯甲烷洗涤两次,随后将滤饼分散于50mL二氯甲烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得34.1g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为92.6%,产率为83.1%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
实施例3
一种4-溴-2-氟苯胺的选择性合成方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入6.1g(0.022mol)四丁基氯化铵、22.2g(0.20mol)邻氟苯胺和50mL二氯甲烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至回流(约40℃)反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用20×2mL的二氯甲烷洗涤两次,随后将滤饼分散于50mL二氯甲烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得34.6g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为87.3%,产率为79.5%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
实施例4
一种4-溴-2-氟苯胺的选择性合成方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入3.65g(0.022mol)四乙基氯化铵、22.2g(0.20mol)邻氟苯胺和50mL二氯甲烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至回流(约40℃)反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用20×2mL的二氯甲烷洗涤两次,随后将滤饼分散于50mL二氯甲烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得33.9g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为85.2%,产率为76.0%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
实施例5
一种4-溴-2-氟苯胺的选择性合成方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入7.1g(0.022mol)四丁基溴化铵、22.2g(0.20mol)邻氟苯胺和50mL环己烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至约40℃反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用20×2mL的环己烷洗涤两次,随后将滤饼分散于50mL环己烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得34.5g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为93.5%,产率为84.9%。
按照实施例1中的表征方式对本实施例的产品进行检测,证明本实施例得到的产物为目标产物。
比较例
直接用液溴溴代邻氟苯胺合成4-溴-2-氟的方法,包括以下步骤:
1)合成:向250mL三口烧瓶中加入22.2g(0.20mol)邻氟苯胺和50mL二氯甲烷,搅拌至全部溶解,在控制温度低于30℃的条件下,缓慢滴加33.6g(0.21mol)溴素,约30~60min滴完,之后将物料温度升至回流(约40℃)反应约2.5h;
2)后处理:冷却至室温,过滤,滤饼用20×2mL的二氯甲烷洗涤两次,随后将滤饼分散于50mL二氯甲烷中,再加约80g 10%的氢氧化钠溶液中和至pH=7~8,静置分出水相,有机相用50×2mL的去离子水洗涤两次后蒸出溶剂得34.2g(理论量38g)4-溴-2-氟苯胺。气相色谱分析表明,产品含量为63.5%,产率为57.2%。
和实施例1相比,比较例的产品含量和产率均低近30%。
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!