一种枸橼酸托法替布晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,涉及一种枸橼酸托法替布晶型化合物及其制备方法。
背景技术:
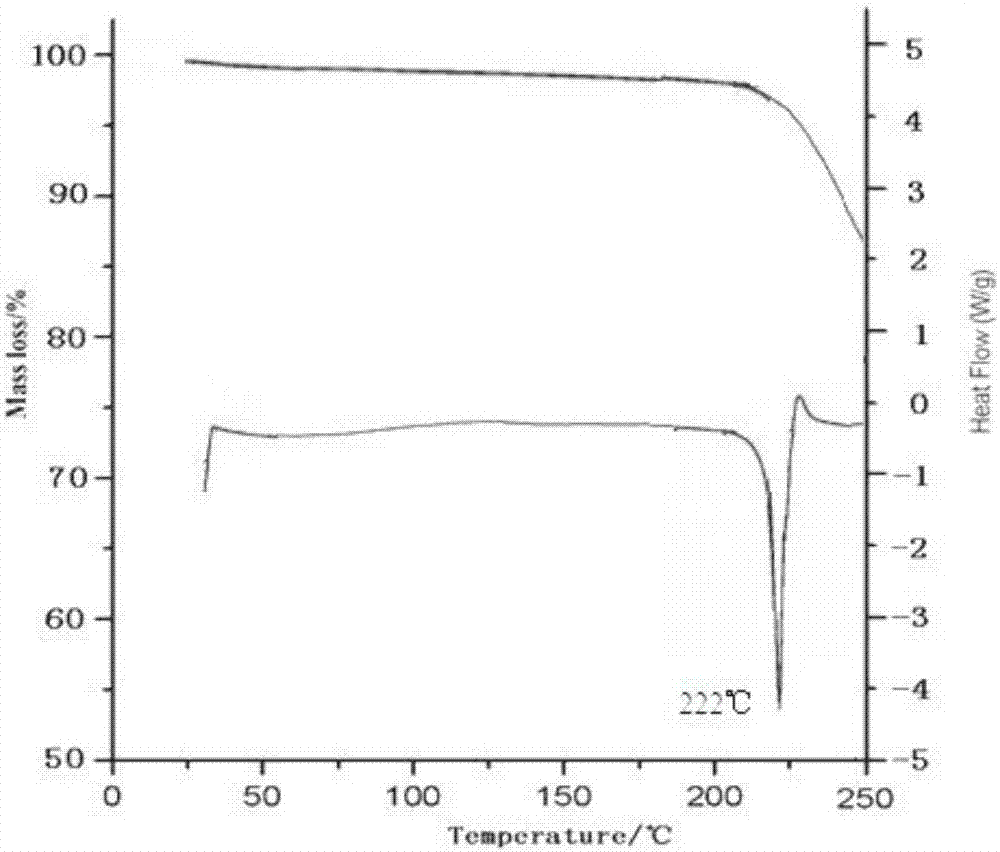
:美国食品药品管理局2012年11月批准了辉瑞的xeljanz(tofacitinib,枸橼酸托法替布),用于对氨甲喋呤治疗反应不足或不耐受的中度至重度活动性类风湿性关节炎(ra)成人患者的治疗。ra是一种自身免疫疾病,是由于免疫系统错误地攻击,导致关节及周围组织产生炎症。据cdc统计,在美国约有150万人受ra困扰。枸橼酸托法替尼上市后将与雅培的重磅药物阿达木单抗(humira)形成竞争,该药被业内人士预测为潜在的重磅药物。柠檬酸托法替布,化学名称为:3-[(3r,4r)-4-甲基-3-[甲基-(7h-吡咯并[2,3-d]嘧啶-4-基)-氨基]-哌啶-1-基]-3-氧代-丙腈柠檬酸盐,分子式:c16h20n6o·c6h8o7;分子量:504.5;本品在二甲基乙酰胺中易溶,在水中微溶,在乙醇中极微溶。本品为白色至白色粉末,pka值为5.1,其在0.1n的盐酸溶液、ph4.5、ph7.4、ph9.0的水溶液中溶解度分别大于10mg/ml、2.4mg/ml、3.5mg/ml和2.3mg/ml。其在乙醇、甲醇、乙腈、丙酮中的溶解度分别为0.6mg/ml、1.6mg/ml、0.07mg/ml和0.3mg/ml。熔程为193.8~195.3℃。结构如式(i)所示:pct申请wo2003048162公开了柠檬酸托法替布一种新晶型及其制备方法,但根据其方法制备得到的晶体颗粒粒径较大,并且在放置一段时间后其颜色会变深,因此,wo2003048162公开的晶体颗粒不稳定,不能满足制剂中对活性成分的要求。所述枸橼酸托法替布成品的x衍射粉末衍射图中的特征峰在2θ=5.76,14.82,16.02,17.43,18.72,20.16,20.48,21.10,21.99,27.00±0.1°。专利201480054229.1提供了中值粒径小于约40μm的托法替布柠檬酸盐的晶体颗粒,具有良好的流动性,可压缩性和可成形性,适于在制剂制备过程中直接压片,因而可避免制粒步骤。还提供的包含中值粒径小于约40μm的托法替布柠檬酸盐的晶体颗粒的药物组合物,在体外可具有良好的溶出速率,在不同的溶出介质中,在5分钟内的平均溶出量可达75%以上,在30分钟内的平均溶出量可达90%以上。因此,在本发明提供的药物组合物可具有更好的溶出和溶解性能。一个原料药的不同多晶型可以有不同的化学和物理特性,包括熔点、化学反应性、表观溶解度、溶解速率、光学和机械性质、蒸气压和密度。这些特性可以直接影响原料药和制剂的处理和/或生产,并且会影响制剂的稳定性、溶解度和生物利用度。当化合物存在多晶型时,由于特定多晶型物具有特异性的热力学性质和稳定性,因此在制备的过程中,了解在各个剂型中应用的化合物的晶型是重要的,以保证生产过程应用相同形态的药物活性化合物。因此,保持药物活性化合物是单一的晶型或是一些晶型的已知混合物是必要的。技术实现要素:本发明的首要发明目的在于提出了一种枸橼酸托法替布晶型化合物及其制备方法。为了实现本发明的目的,采用的技术方案为:本发明提供一种枸橼酸托法替布晶型化合物,该化合物以2θ±0.2°衍射角表示的x-射线粉末衍射图谱在2.42°、3.25°、4.23°、5.04°、6.12°、7.08°、8.02°、9.72°、10.63°、12.90°、14.35°、15.71°、17.82°、18.34°、19.24°、22.46°、24.56°、26.62°、30.75°和32.45°处显示有特征衍射峰。本发明提供的枸橼酸托法替布晶型化合物使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示。本发明还提供了一种枸橼酸托法替布晶型化合物的制备方法,具体步骤为:a)将枸橼酸托法替布粗品溶于混合溶剂a,溶液搅拌加热后使枸橼酸托法替布粗品全部溶解,至溶液澄清,趁热过滤;b)将上述得到的溶液降温,当降至20~30℃时向溶液中按1.0~2.0ml/min的流速加入预冷的混合溶剂b至出晶,析出晶体,继续降温至-5℃~0℃,保温搅拌养晶至析晶完全;c)抽滤,收集晶体,少量乙醇洗涤,真空干燥,得到枸橼酸托法替布结晶。优选地,步骤a)中,所述混合溶剂a为水和甲醇的混合溶剂,水和甲醇的体积比为2~3:1;枸橼酸托法替布和混合溶剂a的质量体积比为1:10~20。优选地,步骤b)中,所述的混合溶剂b为乙醇和丙酮的混合溶剂,乙醇和丙酮的体积比为1:3~5;混合溶剂a与混合溶剂b的体积比为1:2~5。更优选地,步骤b)中,降温幅度为每10分钟1℃~2℃,养晶温度为-5℃~0℃,养晶时间为4~12h。本发明还提供了一种含有本发明枸橼酸托法替布晶型化合物的药物组合物,该药物组合物为含有枸橼酸托法替布晶体的片剂。研究表明,在x-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本发明所提供的枸橼酸托法替布结晶其x-射线粉末衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。下面通过对本发明提供的枸橼酸托法替布晶型化合物进行研究来解释和说明本发明技术方案:1、晶型检测取本发明制备得到的枸橼酸托法替布结晶,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图在2.42°、3.25°、4.23°、5.04°、6.12°、7.08°、8.02°、9.72°、10.63°、12.90°、14.35°、15.71°、17.82°、18.34°、19.24°、22.46°、24.56°、26.62°、30.75°和32.45°处显示有特征峰。2、差热分析及热重分析对本发明制备的枸橼酸托法替布晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在200℃前没有吸收峰,说明样品中无结晶水或结晶溶剂;本品在222℃处有吸热峰。本品经熔点测定:221~223℃,从侧面证明了其为一种不同的晶型。3、水分分析采用卡式水分测定仪测定,本发明的枸橼酸托法替布结晶的含水量为0.07%。4、纯度检测经hplc纯度检测,本发明制备得到的枸橼酸托法替布结晶的纯度可达到99.7~99.9%。与现有技术相比,本发明具有如下优点:(1)本发明所提供的枸橼酸托法替布晶型化合物是一种不同于现有技术的新晶型;(2)本发明所提供的枸橼酸托法替布晶型化合物溶解性性得到了改善,稳定性、流动性好,很好地改善了枸橼酸托法替布在水中的溶解度,提高了生物利用度,有助于药物给药途径的选择设计和药物制剂工艺参数的确定,从而提高药品生产质量;(3)本发明所提供的枸橼酸托法替布晶型化合物制备方法简单易操作,反应条件温和,适合大规模生产。附图说明图1为本发明实施例1制备的枸橼酸托法替布晶型化合物的x-射线粉末衍射图谱。图2本发明实施例1制备的枸橼酸托法替布晶型化合物的tg-dsc图谱。具体实施例以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例1:枸橼酸托法替布晶型化合物的制备取枸橼酸托法替布100g于反应瓶中,加入1000ml水和乙醇的混合溶液(水和乙醇的体积比为3:1),加热至70℃,搅拌溶清,趁热过滤;边搅拌,边降温至20℃(降温幅度为每10分钟2℃),向溶液中按1.0ml/min的流速加入预冷的混合溶剂b(乙醇和丙酮的体积比为1:3)2000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌3h,养晶4h。真空抽滤,滤饼于50℃真空干燥6h,得90.6g白色固体。实施例2:枸橼酸托法替布晶型化合物的制备取枸橼酸托法替布100g于反应瓶中,加入1500ml水和乙醇的混合溶液(水和乙醇的体积比为2:1),加热至60℃,搅拌溶清,趁热过滤;边搅拌,边降温至25℃(降温幅度为每10分钟2℃),向溶液中按1.5ml/min的流速加入预冷的混合溶剂b(乙醇和丙酮的体积比为1:5)3000ml至出晶,继续降温至-10℃(降温幅度为每10分钟2℃),搅拌2h,养晶8h。真空抽滤,滤饼于50℃真空干燥4h,得89.4g白色固体。所制得晶体的使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例3:枸橼酸托法替布晶型化合物的制备取枸橼酸托法替布150g于反应瓶中,加入1500ml水和乙醇的混合溶液(水和乙醇的体积比为2:1),加热至65℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟1℃),向溶液中按2ml/min的流速加入预冷的混合溶剂b(乙醇和丙酮的体积比为1:5)3000ml至出晶,继续降温至0℃(降温幅度为每10分钟1℃),搅拌2h,养晶6h。真空抽滤,滤饼于50℃真空干燥5h,得138g白色固体。所制得的枸橼酸托法替布晶体使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例4:枸橼酸托法替布晶型化合物的制备取枸橼酸托法替布80g于反应瓶中,加入800ml水和乙醇的混合溶液(水和乙醇的体积比为3:1),加热至65℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟1℃),向溶液中按2ml/min的流速加入预冷的混合溶剂b(乙醇和丙酮的体积比为1:4)4000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌2h,养晶10h。真空抽滤,滤饼于50℃真空干燥5h,得74.6g白色固体。所制得的枸橼酸托法替布晶体使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。实施例5:枸橼酸托法替布晶型化合物的制备取枸橼酸托法替布80g于反应瓶中,加入1600ml水和乙醇的混合溶液(水和乙醇的体积比为2:1),加热至70℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟1℃),向溶液中按2ml/min的流速加入预冷的混合溶剂b(乙醇和丙酮的体积比为1:5)3200ml至出晶,继续降温至0℃(降温幅度为每10分钟2℃),搅拌2h,养晶12h。真空抽滤,滤饼于50℃真空干燥5h,得73.5g白色固体。所制得的枸橼酸托法替布晶体使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。下面通过实验例进一步说明本发明:实验例1:流动性实验本实验例采用固定漏斗法测定各实施例样品的休止角,从而评价本发明提供的枸橼酸托法替布结晶的流动性。具体方法如下:将漏斗置于坐标纸上的适宜高度,取实施例1-5批制备的样品,从固定的漏斗中自由留下,直到形成的圆锥体顶部与漏斗口接触,测算物料堆积层的斜边与水平线的夹角度数(休止角θ)。实验结果如表1所示。表1:流动性实验结果样品12345平均值θ(°)33.434.333.634.633.733.9从表1的实验结果分析,本发明实施例1-5制备得到的枸橼酸托法替布结晶的流动性很好,有利于提高分装的准确性,并且与其他成分混合时易于混合均匀。实验例2:溶解度测定试验品:本发明实施例1-5所制备的样品;对照品1是市售枸橼酸托法替布原料药;对照品2是参照专利cn201210349476.7实施例2制备的枸橼酸托法替布晶型;对照品3是参照专利cn201410003888.4实施例2制备的枸橼酸托法替布晶型;参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表2。表2本发明的晶型和对照品在水中溶解性试验结果将上述实施例1-3溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μl测定药物含量即为水中溶解度(mg/ml)。结果见表3:表3本发明晶型与现有技术晶型在水中的溶解度对比从表2-3可以看出,25℃下,本发明枸橼酸托法替布晶体化合物的在水中的溶解度与现有技术相比,有显著提高。实验例3:稳定性试验本实验例通过加速试验和长期试验,考察本发明提供的枸橼酸托法替布结晶的稳定性。1、加速试验取实施例1-3制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、含量,结果见表4。表4:加速试验结果(温度40±2℃,相对湿度75±5%)从表4看出,本发明枸橼酸托法替布结晶在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量没有明显升高,各指标均无明显变化,说明本品稳定性好。2、长期试验取实施例1-3制备的样品,于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、含量,结果见表5。表5:长期试验结果(温度25±2℃,相对湿度60±5%)从表5看出,本发明枸橼酸托法替布结晶在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1