聚酰亚胺膜及覆铜层叠板的制作方法
本发明涉及一种聚酰亚胺膜及覆铜层叠板。
背景技术:
近年来,随着电子设备的小型化、轻量化、省空间化的发展,薄且轻量、具有可挠性、即便反复弯曲也具有优异耐久性的柔性印刷布线板(flexibleprintedcircuits,fpc)的需要不断增大。fpc即便在有限空间内也可实现立体且高密度的安装,因此其用途不断地扩大到例如硬盘驱动器(harddiskdrive,hdd)、数字通用光盘(digitalversatiledisc,dvd)、手机等电子设备的可动部分的布线或电缆(cable)、连接器(connector)等零件。
fpc是通过蚀刻覆铜层叠板(copper-cladlaminate,ccl)的铜层进行布线加工而制造。对于手机或智能手机中连续弯曲或弯折180°的fpc,大多使用压延铜箔作为铜层的材料。例如专利文献1中提出:以耐折裂次数来规定使用压延铜箔所制作的覆铜层叠板的耐弯曲性。另外,专利文献2中提出了一种使用以光泽度及弯折次数规定的压延铜箔的覆铜层叠板。
在对覆铜层叠板的光刻(photolithography)工序或安装fpc的过程中,以设置在覆铜层叠板中的对准标记(alignmentmark)为基准而进行接合、切断、曝光、蚀刻等各种加工。这些工序中的加工精度在维持搭载有fpc的电子设备的可靠性方面变得重要。然而,覆铜层叠板具有将热膨胀系数不同的铜层与树脂层加以层叠的结构,因此由于铜层与树脂层的热膨胀系数之差而在层间产生应力。该应力的一部分或全部在蚀刻铜层进行布线加工的情况下被解除,由此产生伸缩,导致布线图案的尺寸变化。因此,最终在fpc的阶段中发生尺寸变化,成为引起布线间或布线与端子的接触不良的原因,使电路基板的可靠性或良率降低。因此,对于作为电路基板材料的覆铜层叠板来说,尺寸稳定性为非常重要的特性。但是,所述专利文献1、专利文献2中,关于覆铜层叠板的尺寸稳定性未作任何考虑。
作为制造聚酰亚胺膜的方法,已知以下方法:对于聚酰胺酸的自支撑性凝胶膜,同时或连续地进行单轴延伸与热酰亚胺化,由此使聚酰亚胺分子链取向而表现出面内双折射。此时,为了控制延迟(retardation),高精度地控制单轴延伸操作及热酰亚胺化时的升温速度、最终固化温度、负重等条件。例如专利文献3中提出了以下技术:将聚酰亚胺膜一面加热一面单轴延伸,由此控制延迟。
[现有技术文献]
[专利文献]
[专利文献1]日本专利特开2014-15674公报(权利要求书等)
[专利文献2]日本专利特开2014-11451号公报(权利要求书等)
[专利文献3]日本专利特开2000-356713号公报(权利要求书等)
技术实现要素:
[发明所要解决的问题]
本发明的第一方面提供一种可减少高温加工时的尺寸变化的聚酰亚胺膜及使用该聚酰亚胺膜的覆铜层叠板。另外,本发明的第二方面提供一种即便置于热塑性聚酰亚胺的玻璃转移温度以上的加热环境下也实现高的尺寸稳定精度,且可稳定地生产的聚酰亚胺膜及使用该聚酰亚胺膜的覆铜层叠板。
[解决问题的技术手段]
本发明人等进行了努力研究,结果发现,通过控制聚酰亚胺膜的面内延迟可解决所述课题,以至完成了本发明。
即,本发明的聚酰亚胺膜在包含非热塑性聚酰亚胺的非热塑性聚酰亚胺层的至少一侧具有包含热塑性聚酰亚胺的热塑性聚酰亚胺层。本发明的聚酰亚胺膜的特征在于满足下述条件(i)~条件(iv);
(i)热膨胀系数为10ppm/k~30ppm/k的范围内;
(ii)所述热塑性聚酰亚胺的玻璃转移温度为200℃以上且350℃以下的范围内;
(iii)面内延迟(ro)的值为5nm以上且50nm以下的范围内;
(iv)宽度方向(transversedirection,td方向)的面内延迟(ro)的不均一性(δro)为10nm以下。
本发明的聚酰亚胺膜也可为在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量为20nm以下。
本发明的聚酰亚胺膜也可使所述非热塑性聚酰亚胺含有四羧酸残基及二胺残基,所述四羧酸残基及二胺残基均为芳香族基,所述芳香族基包含联苯四基或亚联苯基,并且相对于所述四羧酸残基及二胺残基的合计100摩尔份,所述联苯四基或亚联苯基为40摩尔份以上。
本发明的聚酰亚胺膜也可使所述热塑性聚酰亚胺含有四羧酸残基及二胺残基,所述四羧酸残基及二胺残基均为芳香族基,所述芳香族基包含联苯四基或亚联苯基,并且相对于所述四羧酸残基及二胺残基的合计100摩尔份,所述联苯四基或亚联苯基为30摩尔份以上且80摩尔份以下的范围内。
本发明的聚酰亚胺膜也可为相对于所述非热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,由3,3′,4,4′-联苯四羧酸二酐所衍生的四羧酸残基为20摩尔份以上且70摩尔份以下的范围内。
本发明的聚酰亚胺膜也可为相对于所述热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,由3,3′,4,4′-联苯四羧酸二酐所衍生的四羧酸残基为40摩尔份以上。
本发明的聚酰亚胺膜也可为相对于所述非热塑性聚酰亚胺所含的所有二胺残基100摩尔份,下述通式(1)所表示的二胺残基为20摩尔份以上。
[化1]
[式中,r1、r2独立地表示可经卤素原子或苯基取代的碳数1~3的烷基或碳数1~3的烷氧基或者碳数2~3的烯基]
本发明的聚酰亚胺膜也可为相对于所述热塑性聚酰亚胺所含的所有二胺残基100摩尔份,下述通式(2)所表示的二胺残基为3摩尔份以上且60摩尔份以下的范围内。
[化2]
[式中,r3、r4独立地表示可经卤素原子或苯基取代的碳数1~3的烷基或碳数1~3的烷氧基或者烯基]
本发明的覆铜层叠板具备绝缘层及位于该绝缘层的至少一个面上的铜层。而且,本发明的覆铜层叠板的特征在于:所述绝缘层具有与所述铜层的表面接触的热塑性聚酰亚胺层、及间接地层叠的非热塑性聚酰亚胺层,
所述绝缘层包含所述任一种聚酰亚胺膜。
本发明的覆铜层叠板也可为所述铜层的蚀刻前后的长度方向(machinedirection,md方向)的尺寸变化量及宽度方向(td方向)的尺寸变化量均为2%以下。
[发明的效果]
本发明的聚酰亚胺膜即便在高温、高压的环境下延迟的变化量也得到抑制,因此例如即便为在高温下与铜箔热压接的情况,尺寸稳定性也优异。因此,通过使用本发明的聚酰亚胺膜,可缩短覆铜层叠板的制造工序的时间,生产稳定性优异。特别在以卷对卷(roll-to-roll)方式对宽幅的聚酰亚胺膜进行处理,层叠铜箔而制造覆铜层叠板的情况下,膜的全宽也尺寸变化率低,尺寸稳定,因此可将由该覆铜层叠板所得的fpc高密度安装。因此,通过将本发明的聚酰亚胺膜及使用该聚酰亚胺膜的覆铜层叠板用作fpc材料,可对电路基板实现可靠性及良率的提高。
附图说明
图1为表示对本发明的一实施方式的覆铜层叠板的尺寸稳定性进行评价的评价方法中所用的覆铜层叠板与试片的概略构成的立体图。
图2为对试片中的标记位置进行说明的图。
图3为试片的中心区域的局部放大图。
图4为试片的角落区域的局部放大图。
图5为对孔与孔的间隔的尺寸变化量进行说明的图。
图6为供说明实施例的评价样本的图。
图7为供说明实施例的评价样本的制备的图。
[符号的说明]
10:试片
20:假想正四边形
20a:中心
20b:角部
21:中心区域
23a、23b:角落区域
30:孔
30a:孔30的中心
100:覆铜层叠板
l0、l1:距离
md、td:方向
δ1、δ1+δ2:尺寸变化量
具体实施方式
其次,一面适当参照附图一面对本发明的实施方式进行说明。
<聚酰亚胺膜>
本实施方式的聚酰亚胺膜在非热塑性聚酰亚胺层的至少一侧具有热塑性聚酰亚胺层。即,热塑性聚酰亚胺层是设置在非热塑性聚酰亚胺层的单面或两面上。例如在制成由本实施方式的聚酰亚胺膜与铜层所构成的覆铜层叠板的情况下,铜层是层叠在热塑性聚酰亚胺层的面上。
这里所谓非热塑性聚酰亚胺,通常为即便进行加热产生软化而也不显示出粘接性的聚酰亚胺,而本发明中是指使用动态粘弹性测定装置(动态机械分析(dynamicmechanicalanalysis,dma))所测定的30℃下的储存模数为1.0×109pa以上,且360℃下的储存模数为1.0×108pa以上的聚酰亚胺。另外,所谓热塑性聚酰亚胺,通常为可明确地确认到玻璃转移温度(tg)的聚酰亚胺,而本发明中是指使用dma所测定的30℃下的储存模数为1.0×109pa以上,且360℃下的储存模数小于1.0×108pa的聚酰亚胺。
本实施方式的聚酰亚胺膜可为膜(片),也可为层叠在铜箔、玻璃板、聚酰亚胺系膜、聚酰胺系膜、聚酯系膜等树脂片等基材上的状态的膜。
本实施方式的聚酰亚胺膜例如在应用作电路基板的绝缘层的情况下,为了防止翘曲的产生或尺寸稳定性的降低,重要的是热膨胀系数(coefficientsofthermalexpansion,cte)为10ppm/k以上且30ppm/k以下的范围内,优选以10ppm/k以上且25ppm/k以下的范围内为宜。若cte小于10ppm/k或超过30ppm/k,则产生翘曲,或尺寸稳定性降低。另外,本实施方式的聚酰亚胺膜中,相对于包含铜箔等的铜层的cte,聚酰亚胺膜的cte更优选±5ppm/k以下的范围内,最优选±2ppm/k以下的范围内。
本实施方式的聚酰亚胺膜中,聚酰亚胺膜的厚度可根据使用目的而设定为既定范围内的厚度。聚酰亚胺膜的厚度例如优选在8μm~50μm的范围内,更优选在11μm~26μm的范围内。若聚酰亚胺膜的厚度小于所述下限值,则有时无法确保电绝缘性,或产生因操作性降低而在制造工序中处理变困难等问题。另一方面,若聚酰亚胺膜的厚度超过所述上限值,则为了控制面内延迟(ro),必须高精度地控制制造条件,产生生产性降低等不良状况。
本实施方式的聚酰亚胺膜中,非热塑性聚酰亚胺层构成低热膨胀性的聚酰亚胺层,热塑性聚酰亚胺层构成高热膨胀性的聚酰亚胺层。这里,低热膨胀性的聚酰亚胺层是指热膨胀系数(cte)优选1ppm/k以上且25ppm/k以下的范围内、更优选3ppm/k以上且25ppm/k以下的范围内的聚酰亚胺层。另外,高热膨胀性的聚酰亚胺层是指cte优选35ppm/k以上、更优选35ppm/k以上且80ppm/k以下的范围内、进而优选35ppm/k以上且70ppm/k以下的范围内的聚酰亚胺层。聚酰亚胺层可通过适当变更所使用的原料的组合、厚度、干燥及硬化条件而制成具有所需cte的聚酰亚胺层。
另外,本实施方式的聚酰亚胺膜中,以非热塑性聚酰亚胺层与热塑性聚酰亚胺层的厚度比(非热塑性聚酰亚胺层/热塑性聚酰亚胺层)为1.5~6.0的范围内为宜。该比的值若小于1.5,则相对于聚酰亚胺膜总体的非热塑性聚酰亚胺层变薄,因此面内延迟(ro)的不均一性容易变大,若超过6.0则热塑性聚酰亚胺层变薄,因此聚酰亚胺膜与铜层的粘接可靠性容易降低。该面内延迟(ro)的控制与构成聚酰亚胺膜的各聚酰亚胺层的树脂构成及其厚度有关。对于作为赋予粘接性即高热膨胀性或软化的树脂构成的热塑性聚酰亚胺层来说,其厚度越大,则对聚酰亚胺膜的ro的值造成的影响越大。因此,增大非热塑性聚酰亚胺层的厚度的比率,减小热塑性聚酰亚胺层的厚度的比率,减小聚酰亚胺膜的ro的值及其不均一性。
本实施方式的聚酰亚胺膜中,构成热塑性聚酰亚胺层的聚酰亚胺可提高与铜层的密接性。这种热塑性聚酰亚胺的玻璃转移温度为200℃以上且350℃以下的范围内,优选200℃以上且320℃以下的范围内。
就更大地表现出本实施方式的聚酰亚胺膜的尺寸精度的改善效果的观点来看,本实施方式的聚酰亚胺膜优选的是膜宽为490mm以上且1100mm以下的范围内,且长条状的长度为20m以上。在连续地制造本实施方式的聚酰亚胺膜的情况下,宽度方向(以下也称为td方向)越宽的膜,发明的效果越尤其变明显。此外,也包括连续地制造本实施方式的聚酰亚胺膜后,在长条的聚酰亚胺膜的长度方向(以下也称为md方向)及td方向上以某一定的值切割(slit)所得的膜。
本实施方式的聚酰亚胺膜的面内延迟(ro)的值为5nm以上且50nm以下的范围内,优选5nm以上且20nm以下的范围内,更优选5nm以上且15nm以下的范围内。另外,td方向的ro的不均一性(δro)为10nm以下,优选5nm以下,更优选3nm以下,由于在这种范围内进行控制,因此尤其即便是厚度为25μm以上的膜,尺寸精度也提高。
本实施方式的聚酰亚胺膜优选的是在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量为20nm以下,更优选10nm以下,进而优选5nm以下。本实施方式的聚酰亚胺膜即便在超过构成热塑性聚酰亚胺层的聚酰亚胺的玻璃转移温度的温度下,也将ro的变化量控制于所述上限值以下。因此,例如在通过热层压将本实施方式的聚酰亚胺膜与铜箔贴合的工序前后,ro也不易变化,因此成为尺寸稳定性优异的聚酰亚胺膜。
另外,本实施方式的聚酰亚胺膜的拉伸模数优选3.0gpa~10.0gpa的范围内,更优选4.5gpa~8.0gpa的范围内。若聚酰亚胺膜的拉伸模数小于3.0gpa,则聚酰亚胺自身的强度降低,由此将覆铜层叠板加工成电路基板时有时产生膜的开裂等操作上的问题。相反地,若聚酰亚胺膜的拉伸模数超过10.0gpa,则覆铜层叠板的对于弯折的刚性上升,结果在将覆铜层叠板弯折时,对铜布线施加的弯曲应力上升,耐弯折性降低。通过将聚酰亚胺膜的拉伸模数设为所述范围内,可确保聚酰亚胺膜的强度及柔软性。
本实施方式的聚酰亚胺膜的制造方法的实施方式例如有:[1]在支撑基材上涂布聚酰胺酸的溶液并进行干燥后,加以酰亚胺化而制造聚酰亚胺膜的方法;以及[2]在支撑基材上涂布聚酰胺酸的溶液并进行干燥后,将聚酰胺酸的凝胶膜从支撑基材剥离,加以酰亚胺化而制造聚酰亚胺膜的方法。另外,本实施方式的聚酰亚胺膜为包含多层聚酰亚胺层的聚酰亚胺膜,因此其制造方法的实施方式例如可举出:[3]在支撑基材上重复进行聚酰胺酸的溶液的涂布、干燥后,进行酰亚胺化的方法(以下称为浇注法);以及[4]通过多层挤出同时将聚酰胺酸以多层地层叠的状态进行涂布、干燥后,进行酰亚胺化的方法(以下称为多层挤出法)等。
所述[1]方法例如可包括以下的工序1a~工序1c;
(1a)在支撑基材上涂布聚酰胺酸的溶液,并加以干燥的工序;
(1b)在支撑基材上对聚酰胺酸进行热处理而酰亚胺化,由此形成聚酰亚胺层的工序;以及
(1c)将支撑基材与聚酰亚胺层分离,由此获得聚酰亚胺膜的工序。
所述[2]方法例如可包括以下的工序2a~工序2c;
(2a)在支撑基材上涂布聚酰胺酸的溶液,并加以干燥的工序;
(2b)将支撑基材与聚酰胺酸的凝胶膜分离的工序;以及
(2c)对聚酰胺酸的凝胶膜进行热处理而酰亚胺化,由此获得聚酰亚胺膜的工序。
所述[3]方法是在所述[1]方法或[2]方法中重复进行多次工序1a或工序2a,在支撑基材上形成聚酰胺酸的层叠结构体,除此以外,可与所述[1]方法或[2]方法同样地实施。
所述[4]方法是在所述[1]方法的工序1a或[2]方法的工序2a中,通过多层挤出同时涂布聚酰胺酸的层叠结构体并进行干燥,除此以外,可与所述[1]方法或[2]方法同样地实施。
本发明中制造的聚酰亚胺膜优选的是在支撑基材上完成聚酰胺酸的酰亚胺化。由于在将聚酰胺酸的树脂层固定在支撑基材上的状态下进行酰亚胺化,因此可抑制酰亚胺化过程中的聚酰亚胺层的伸缩变化,维持聚酰亚胺膜的厚度或尺寸精度。
然而,对于在支撑基材上完成聚酰胺酸的酰亚胺化的聚酰亚胺膜来说,由于在从支撑基材分离聚酰亚胺膜时所施加的对聚酰亚胺膜的张力、或例如在使用刀锋(knifeedge)等的剥离时所产生的对聚酰亚胺膜的应力等,而将聚酰亚胺膜延伸。因此,容易产生聚酰亚胺膜的面内延迟(ro)的不均一性,尤其越是膜宽为490mm以上的聚酰亚胺膜,ro的不均一性越变明显。对于本实施方式的聚酰亚胺膜来说,以构成非热塑性聚酰亚胺层及热塑性聚酰亚胺层的聚酰亚胺均容易形成秩序结构的方式进行设定,由此使剥离所必需的应力分散至聚酰亚胺膜的各层中,由此可控制ro。
另外,也可通过以下方法来控制面内延迟(ro):将支撑基材上的聚酰胺酸的凝胶膜分离,将聚酰胺酸的凝胶膜单轴延伸或双轴延伸,同时或连续地进行酰亚胺化。此时,为了更精密地高度地控制ro,优选的是适当调整延伸操作及酰亚胺化时的升温速度、酰亚胺化的完成温度、负重等条件。
(非热塑性聚酰亚胺)
本实施方式的聚酰亚胺膜中,优选的是非热塑性聚酰亚胺含有四羧酸残基及二胺残基,这些残基均为芳香族基,且包含联苯四基或亚联苯基。这里,联苯四基或亚联苯基与二苯基骨架为相同含意,例如也可使卤素原子、烷基、烷氧基、烯基等取代基键合于联苯四基或亚联苯基,但尤其从减小高温环境下的面内延迟(ro)的变化量的观点来看,例如更优选的是将烷基、烷氧基、烯基等取代基的碳数设为1~3的范围内。
此外,本发明中,所谓四羧酸残基,表示由四羧酸二酐所衍生的四价基团,所谓二胺残基,表示由二胺化合物所衍生的二价基团。另外,二胺化合物为具有两个氨基的化合物,但各氨基中的氢原子也可经任意的取代基所取代。
非热塑性聚酰亚胺所含的四羧酸残基及二胺残基均为芳香族基,通过设为芳香族基,可减小聚酰亚胺膜的高温环境下的面内延迟(ro)的变化量。进而,相对于四羧酸残基及二胺残基的合计100摩尔份,优选的是将联苯四基或亚联苯基设为40摩尔份以上,更优选的是设为50摩尔份以上,由此容易形成由联苯四基或亚联苯基所得的秩序结构,可减小聚酰亚胺膜的高温环境下的ro的变化量,并且抑制ro的不均一性。
另外,非热塑性聚酰亚胺所含的四羧酸残基例如可优选地举出由3,3′,4,4′-联苯四羧酸二酐(biphenyltetracarboxylicdianhydride,bpda)、2,2′,3,3′-联苯四羧酸二酐等所衍生的四羧酸残基。这些四羧酸残基中,尤其由bpda所衍生的四羧酸残基(以下也称为bpda残基)容易形成秩序结构,可减小高温环境下的面内延迟(ro)的变化量,因此特别优选。另外,bpda残基虽可赋予作为聚酰亚胺前驱物的聚酰胺酸的凝胶膜的自支撑性,但有使酰亚胺化后的cte增大的倾向。从这种观点来看,相对于非热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,bpda残基以优选20摩尔份~70摩尔份的范围内、更优选20摩尔份~60摩尔份的范围内为宜。
非热塑性聚酰亚胺所含的所述bpda残基以外的四羧酸残基可优选地举出由均苯四甲酸二酐(pyromelliticdianhydride,pmda)所衍生的四羧酸残基(以下也称为pmda残基)。相对于非热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,pmda残基以优选0摩尔份~60摩尔份的范围内、更优选0摩尔份~50摩尔份的范围内为宜。pmda残基为任意,但为发挥控制热膨胀系数及控制玻璃转移温度的作用的残基。
其他四羧酸残基例如可举出由以下的芳香族四羧酸二酐所衍生的四羧酸残基:3,3′,4,4′-二苯基砜四羧酸二酐、4,4′-氧基双邻苯二甲酸酐、2,3′,3,4′-联苯四羧酸二酐、2,2′,3,3′-二苯甲酮四羧酸二酐、2,3,3′,4′-二苯甲酮四羧酸二酐或3,3′,4,4′-二苯甲酮四羧酸二酐、2,3′,3,4′-二苯基醚四羧酸二酐、双(2,3-二羧基苯基)醚二酐、3,3″,4,4″-对三联苯四羧酸二酐、2,3,3″,4″-对三联苯四羧酸二酐或2,2″,3,3″-对三联苯四羧酸二酐、2,2-双(2,3-二羧基苯基)-丙烷二酐或2,2-双(3,4-二羧基苯基)-丙烷二酐、双(2,3-二羧基苯基)甲烷二酐或双(3,4-二羧基苯基)甲烷二酐、双(2,3-二羧基苯基)砜二酐或双(3,4-二羧基苯基)砜二酐、1,1-双(2,3-二羧基苯基)乙烷二酐或1,1-双(3,4-二羧基苯基)乙烷二酐、1,2,7,8-菲-四羧酸二酐、1,2,6,7-菲-四羧酸二酐或1,2,9,10-菲-四羧酸二酐、2,3,6,7-蒽四羧酸二酐、2,2-双(3,4-二羧基苯基)四氟丙烷二酐、2,3,5,6-环己烷二酐、1,2,5,6-萘四羧酸二酐、1,4,5,8-萘四羧酸二酐、2,3,6,7-萘四羧酸二酐、4,8-二甲基-1,2,3,5,6,7-六氢萘-1,2,5,6-四羧酸二酐、2,6-或2,7-二氯萘-1,4,5,8-四羧酸二酐、2,3,6,7-四氯萘-1,4,5,8-四羧酸二酐或1,4,5,8-四氯萘-2,3,6,7-四羧酸二酐、2,3,8,9-苝-四羧酸二酐、3,4,9,10-苝-四羧酸二酐、4,5,10,11-苝-四羧酸二酐或5,6,11,12-苝-四羧酸二酐、环戊烷-1,2,3,4-四羧酸二酐、吡嗪-2,3,5,6-四羧酸二酐、吡咯烷-2,3,4,5-四羧酸二酐、噻吩-2,3,4,5-四羧酸二酐、4,4′-双(2,3-二羧基苯氧基)二苯基甲烷二酐等。
非热塑性聚酰亚胺所含的四羧酸残基中,相对于非热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,由2,3′,3,4′-二苯基醚四羧酸二酐、3,3′,4,4′-二苯基砜四羧酸二酐、4,4′-氧基双邻苯二甲酸酐及2,3′,3,4′-二苯基四羧酸二酐的四羧酸二酐所衍生的四羧酸残基以优选20摩尔份以下、更优选15摩尔份以下为宜。若相对于非热塑性聚酰亚胺所含的所有四羧酸残基而这些四羧酸残基超过20摩尔份,则分子的取向性降低,面内延迟(ro)的控制变困难。
本实施方式的聚酰亚胺膜中,非热塑性聚酰亚胺所含的二胺残基例如可优选地举出下述通式(1)所表示的二胺残基。
[化3]
所述式(1)中,r1、r2独立地表示可经卤素原子或苯基取代的碳数1~3的烷基、或碳数1~3的烷氧基或者碳数2~3的烯基。
通式(1)所表示的二胺残基容易形成秩序结构,尤其可有利地抑制高温环境下的面内延迟(ro)的变化量。从这种观点来看,相对于非热塑性聚酰亚胺所含的所有二胺残基100摩尔份,通式(1)所表示的二胺残基以优选20摩尔份以上、更优选50摩尔份以上、进而优选60摩尔份~90摩尔份的范围内为宜。
通式(1)所表示的二胺残基的优选具体例可举出由以下的二胺化合物所衍生的二胺残基:2,2′-二甲基-4,4′-二氨基联苯(m-tb)、2,2′-二乙基-4,4′-二氨基联苯(m-eb)、2,2′-二乙氧基-4,4′-二氨基联苯(m-eob)、2,2′-二丙氧基-4,4′-二氨基联苯(m-pob)、2,2′-正丙基-4,4′-二氨基联苯(m-npb)、2,2′-二乙烯基-4,4′-二氨基联苯(vab)、4,4′-二氨基联苯、4,4′-二氨基-2,2′-双(三氟甲基)联苯(tfmb)等。这些二胺化合物中,尤其2,2′-二甲基-4,4′-二氨基联苯(m-tb)容易形成秩序结构,可减小高温环境下的面内延迟(ro)的变化量,因此特别优选。
通式(1)所表示的二胺残基以外的二胺残基可优选地举出由对苯二胺(p-pda)、间苯二胺(m-pda)等所衍生的二胺残基,更优选以由p-pda所衍生的二胺残基(以下也称为pda残基)为宜。相对于非热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,pda残基以优选0摩尔份~80摩尔份的范围内、更优选0摩尔份~50摩尔份的范围内为宜。pda残基为任意,但为发挥控制热膨胀系数及控制玻璃转移温度的作用的残基。
另外,为了提高制成聚酰亚胺膜的情况下的伸长率及耐弯折性等,优选的是非热塑性聚酰亚胺含有选自由下述通式(3)~通式(5)所表示的二胺残基所组成的组群中的至少一种二胺残基。
[化4]
所述式(3)中,r5及r6分别独立地表示氢原子、或卤素原子、或者碳数1~4的可经卤素原子取代的烷基或烷氧基或烯基,x独立地表示选自-o-、-s-、-ch2-、-ch(ch3)-、-c(ch3)2-、-co-、-coo-、-so2-、-nh-或-nhco-中的二价基团,m及n独立地表示1~4的整数。
[化5]
所述式(4)中,r5、r6及r7分别独立地表示氢原子、或卤素原子、或者碳数1~4的可经卤素原子取代的烷基或烷氧基或烯基,x独立地表示选自-o-、-s-、-ch2-、-ch(ch3)-、-c(ch3)2-、-co-、-coo-、-so2-、-nh-或-nhco-中的二价基团,m、n及o独立地表示1~4的整数。
[化6]
所述式(5)中,r5、r6、r7及r8分别独立地表示氢原子、或卤素原子、或者碳数1~4的可经卤素原子取代的烷基或烷氧基或烯基,x1及x2分别独立地表示单键或选自-o-、-s-、-ch2-、-ch(ch3)-、-c(ch3)2-、-co-、-coo-、-so2-、-nh-或-nhco-中的二价基团,将x1及x2两者为单键的情况除外,m、n、o及p独立地表示1~4的整数。
通式(3)~通式(5)所表示的二胺残基具有弯曲性的部位,因此可对聚酰亚胺膜赋予柔软性。此处,通式(4)及通式(5)所表示的二胺残基由于苯环为3个或4个,因此为了抑制热膨胀系数(cte)的增加,优选的是将键合于苯环的末端基设为对位。另外,从对聚酰亚胺膜赋予柔软性并且抑制热膨胀系数(cte)的增加的观点来看,相对于非热塑性聚酰亚胺所含的所有二胺残基100摩尔份,通式(3)~通式(5)所表示的二胺残基以优选10摩尔份~40摩尔份的范围内、更优选10摩尔份~30摩尔份的范围内为宜。若通式(3)~通式(5)所表示的二胺残基小于10摩尔份,则产生制成膜的情况下的伸长率降低,耐弯折性等降低。另一方面,若超过40摩尔份,则分子的取向性降低,低cte化变困难。
通式(3)中,基团r5及基团r6的优选例可举出:氢原子或碳数1~4的可经卤素原子取代的烷基、或者碳数1~3的烷氧基或烯基。另外,通式(3)中,连结基x的优选例可举出-o-、-s-、-ch2-、-ch(ch3)-、-so2-或-co-。通式(3)所表示的二胺残基的优选具体例可举出由以下的二胺化合物所衍生的二胺残基:4,4′-二氨基二苯基醚(4,4′-dape)、3,3′-二氨基二苯基醚、3,4′-二氨基二苯基醚、4,4′-二氨基二苯基甲烷、3,3′-二氨基二苯基甲烷、3,4′-二氨基二苯基甲烷、4,4′-二氨基二苯基丙烷、3,3′-二氨基二苯基丙烷、3,4′-二氨基二苯基丙烷、4,4′-二氨基二苯基硫醚、3,3′-二氨基二苯基硫醚、3,4′-二氨基二苯基硫醚、4,4′-二氨基二苯基砜、3,3′-二氨基二苯基砜、4,4′-二氨基二苯甲酮、3,4′-二氨基二苯甲酮、3,3′-二氨基二苯甲酮等。
通式(4)中,基团r5、基团r6及基团r7的优选例可举出:氢原子或碳数1~4的可经卤素原子取代的烷基、或者碳数1~3的烷氧基或烯基。另外,通式(4)中,连结基x的优选例可举出-o-、-s-、-ch2-、-ch(ch3)-、-so2-或-co-。通式(4)所表示的二胺残基的优选具体例可举出由以下的二胺化合物所衍生的二胺残基:1,3-双(4-氨基苯氧基)苯(tpe-r)、1,4-双(4-氨基苯氧基)苯(tpe-q)、双(4-氨基苯氧基)-2,5-二-叔丁基苯(dtbab)、4,4-双(4-氨基苯氧基)二苯甲酮(bapk)、1,3-双[2-(4-氨基苯基)-2-丙基]苯、1,4-双[2-(4-氨基苯基)-2-丙基]苯等。
通式(5)中,基团r5、基团r6、基团r7及基团r8的优选例可举出:氢原子或碳数1~4的可经卤素原子取代的烷基、或者碳数1~3的烷氧基或烯基。另外,通式(5)中,连结基x1及连结基x2的优选例可举出单键、-o-、-s-、-ch2-、-ch(ch3)-、-so2-或-co-。其中,从赋予弯曲部位的观点来看,将连结基x1及连结基x2两者为单键的情况除外。通式(5)所表示的二胺残基的优选具体例可举出由以下的二胺化合物所衍生的二胺残基:4,4′-双(4-氨基苯氧基)联苯(bapb)、2,2′-双[4-(4-氨基苯氧基)苯基]丙烷(bapp)、2,2′-双[4-(4-氨基苯氧基)苯基]醚(bape)、双[4-(4-氨基苯氧基)苯基]砜等。
其他二胺残基例如可举出由以下的芳香族二胺化合物所衍生的二胺残基:2,2-双-[4-(3-氨基苯氧基)苯基]丙烷、双[4-(3-氨基苯氧基)苯基]砜、双[4-(3-氨基苯氧基)联苯、双[1-(3-氨基苯氧基)]联苯、双[4-(3-氨基苯氧基)苯基]甲烷、双[4-(3-氨基苯氧基)苯基]醚、双[4-(3-氨基苯氧基)]二苯甲酮、9,9-双[4-(3-氨基苯氧基)苯基]芴、2,2-双-[4-(4-氨基苯氧基)苯基]六氟丙烷、2,2-双-[4-(3-氨基苯氧基)苯基]六氟丙烷、3,3′-二甲基-4,4′-二氨基联苯、4,4′-亚甲基二邻甲苯胺、4,4′-亚甲基二-2,6-二甲苯胺、4,4′-亚甲基-2,6-二乙基苯胺、3,3′-二氨基二苯基乙烷、3,3′-二氨基联苯、3,3′-二甲氧基联苯胺、3,3″-二氨基对三联苯、4,4′-[1,4-亚苯基双(1-甲基亚乙基)]双苯胺、4,4′-[1,3-亚苯基双(1-甲基亚乙基)]双苯胺、双(对氨基环己基)甲烷、双(对-β-氨基叔丁基苯基)醚、双(对-β-甲基-δ-氨基戊基)苯、对双(2-甲基-4-氨基戊基)苯、对双(1,1-二甲基-5-氨基戊基)苯、1,5-二氨基萘、2,6-二氨基萘、2,4-双(β-氨基叔丁基)甲苯、2,4-二氨基甲苯、间二甲苯-2,5-二胺、对二甲苯-2,5-二胺、间二甲苯二胺、对二甲苯二胺、2,6-二氨基吡啶、2,5-二氨基吡啶、2,5-二氨基-1,3,4-噁二唑、哌嗪等。
在非热塑性聚酰亚胺中,通过选定所述四羧酸残基及二胺残基的种类、或应用两种以上的四羧酸残基或二胺残基的情况下的各自的摩尔比,可控制热膨胀系数、储存模数、拉伸模数等。另外,在非热塑性聚酰亚胺中具有多种聚酰亚胺的结构单元的情况下,能以嵌段的形式存在,也可无规地存在,从抑制面内延迟(ro)的不均一性的观点来看,优选的是无规地存在。
(热塑性聚酰亚胺)
本实施方式的聚酰亚胺膜中,优选的是热塑性聚酰亚胺含有四羧酸残基及二胺残基,这些残基均为芳香族基,且包含联苯四基或亚联苯基。这里,关于联苯四基或亚联苯基,例如也可使卤素原子、烷基、烷氧基、烯基等取代基键合于联苯四基或亚联苯基,尤其从抑制高温环境下的面内延迟(ro)的变化量的观点来看,例如烷基、烷氧基、烯基等取代基的碳数优选的是设定为1~3的范围内。
热塑性聚酰亚胺所含的四羧酸残基及二胺残基均为芳香族基,通过设为芳香族基,可抑制聚酰亚胺膜的高温环境下的面内延迟(ro)的变化量。进而,相对于四羧酸残基及二胺残基的合计100摩尔份,将联苯四基或亚联苯基设为30摩尔份以上且80摩尔份以下的范围内。若联苯四基或亚联苯基小于30摩尔份,则难以形成由联苯四基或亚联苯基所得的秩序结构,聚酰亚胺膜的高温环境下的ro的变化量增大。另一方面,若联苯四基或亚联苯基超过80摩尔份,则热塑性受损。
另外,热塑性聚酰亚胺所含的四羧酸残基例如可优选地举出由bpda、2,3′,3,4′-联苯四羧酸二酐、2,2′,3,3′-联苯四羧酸二酐等所衍生的四羧酸残基。这些四羧酸残基中,尤其bpda残基容易形成秩序结构,可抑制高温环境下的面内延迟(ro)的变化量,因此特别优选。因此,相对于热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,bpda残基以优选40摩尔份以上、更优选50摩尔份以上为宜。
热塑性聚酰亚胺所含的所述bpda残基以外的四羧酸残基可优选地举出pmda残基。相对于热塑性聚酰亚胺所含的所有四羧酸残基100摩尔份,pmda残基以优选0摩尔份~60摩尔份的范围内、更优选0摩尔份~50摩尔份的范围内为宜。pmda残基为任意,但为发挥控制热膨胀系数及控制玻璃转移温度的作用的残基。
其他四羧酸残基例如可举出由以下的芳香族四羧酸二酐所衍生的四羧酸残基:3,3′,4,4′-二苯基砜四羧酸二酐、4,4′-氧基双邻苯二甲酸酐、2,2′,3,3′-二苯甲酮四羧酸二酐、2,3,3′,4′-二苯甲酮四羧酸二酐或3,3′,4,4′-二苯甲酮四羧酸二酐、2,3′,3,41-二苯基醚四羧酸二酐、双(2,3-二羧基苯基)醚二酐、3,3″,4,4″-对三联苯四羧酸二酐、2,3,3″,4″-对三联苯四羧酸二酐或2,2″,3,3″-对三联苯四羧酸二酐、2,2-双(2,3-二羧基苯基)-丙烷二酐或2,2-双(3,4-二羧基苯基)-丙烷二酐、双(2,3-二羧基苯基)甲烷二酐或双(3,4-二羧基苯基)甲烷二酐、双(2,3-二羧基苯基)砜二酐或双(3,4-二羧基苯基)砜二酐、1,1-双(2,3-二羧基苯基)乙烷二酐或1,1-双(3,4-二羧基苯基)乙烷二酐、1,2,7,8-菲-四羧酸二酐、1,2,6,7-菲-四羧酸二酐或1,2,9,10-菲-四羧酸二酐、2,3,6,7-蒽四羧酸二酐、2,2-双(3,4-二羧基苯基)四氟丙烷二酐、2,3,5,6-环己烷二酐、1,2,5,6-萘四羧酸二酐、1,4,5,8-萘四羧酸二酐、2,3,6,7-萘四羧酸二酐、4,8-二甲基-1,2,3,5,6,7-六氢萘-1,2,5,6-四羧酸二酐、2,6-或2,7-二氯萘-1,4,5,8-四羧酸二酐、2,3,6,7-四氯萘-1,4,5,8-四羧酸二酐或1,4,5,8-四氯萘-2,3,6,7-四羧酸二酐、2,3,8,9-苝-四羧酸二酐、3,4,9,10-苝-四羧酸二酐、4,5,10,11-苝-四羧酸二酐或5,6,11,12-苝-四羧酸二酐、环戊烷-1,2,3,4-四羧酸二酐、吡嗪-2,3,5,6-四羧酸二酐、吡咯烷-2,3,4,5-四羧酸二酐、噻吩-2,3,4,5-四羧酸二酐、4,4′-双(2,3-二羧基苯氧基)二苯基甲烷二酐、2,2-双[4-(3,4-二羧基苯氧基)苯基]丙烷二酐等。
本实施方式的聚酰亚胺膜中,热塑性聚酰亚胺所含的二胺残基例如可优选地举出下述通式(2)所表示的二胺残基。
[化7]
所述式(2)中,r3、r4独立地表示可经卤素原子或苯基取代的碳数1~3的烷基或碳数1~3的烷氧基或者烯基。
通式(2)所表示的二胺残基容易形成秩序结构,尤其可有利地抑制高温环境下的面内延迟(ro)的变化量。从这种观点来看,相对于热塑性聚酰亚胺所含的所有二胺残基100摩尔份,通式(2)所表示的二胺残基以优选3摩尔份~60摩尔份的范围内、更优选5摩尔份~40摩尔份的范围内为宜。若通式(2)所表示的二胺残基小于3摩尔份,则难以形成秩序结构,聚酰亚胺膜的高温环境下的面内延迟(ro)的变化量增大,若超过60摩尔份,则热塑性受损。
通式(2)所表示的二胺残基的优选具体例可举出由以下的二胺化合物所衍生的二胺残基:2,2′-二甲基-4,4′-二氨基联苯(m-tb)、2,2′-二乙基-4,4′-二氨基联苯(m-eb)、2,2′-二乙氧基-4,4′-二氨基联苯(m-eob)、2,2′-二丙氧基-4,4′-二氨基联苯(m-pob)、2,2′-正丙基-4,4′-二氨基联苯(m-npb)、2,2′-二乙烯基-4,4′-二氨基联苯(vab)、4,4′-二氨基联苯、4,4′-二氨基-2,2′-双(三氟甲基)联苯(tfmb)等。这些化合物中,尤其2,2′-二甲基-4,4′-二氨基联苯(m-tb)容易形成秩序结构,可减小高温环境下的面内延迟(ro)的变化量,因此特别优选。
通式(2)所表示的二胺残基以外的二胺残基可优选地举出由对苯二胺(p-pda)、间苯二胺(m-pda)等所衍生的二胺残基,更优选的是以由p-pda所衍生的二胺残基(以下也称为pda残基)为宜。相对于热塑性聚酰亚胺所含的所有二胺残基100摩尔份,pda残基以优选3摩尔份~60摩尔份的范围内、更优选5摩尔份~40摩尔份的范围内为宜。pda残基为任意,但由于具有刚直结构,因此具有对聚合物总体赋予秩序结构的作用。
另外,为了提高聚酰亚胺分子链的柔软性,赋予热塑性,热塑性聚酰亚胺优选的是含有选自由下述通式(6)~通式(12)所表示的二胺残基所组成的组群中的至少一种二胺残基。
[化8]
所述式(6)~式(12)中,r9独立地表示碳数1~6的一价烃基或烷氧基,连结基a独立地表示选自-o-、-s-、-co-、-so-、-so2-、-coo、-ch2-、-c(ch3)2-、-nh-或-conh-中的二价基团,n1独立地表示0~4的整数。其中,从式(8)中将与式(7)重复者除外,从式(10)中将与式(9)重复者除外。此外,所谓“独立地”,是指所述式(6)~式(12)中的一个中或两个以上中,多个连结基a、多个r9或多个n1可相同也可不同。
通式(6)~通式(12)所表示的二胺残基以如下情况为宜:相对于热塑性聚酰亚胺所含的所有二胺残基100摩尔份,至少一种的合计量优选40摩尔份~97摩尔份的范围内。若通式(6)~通式(12)所表示的二胺残基的合计量小于40摩尔份,则聚酰亚胺的柔软性不足而无法获得热塑性,若超过97摩尔份,则有聚酰亚胺膜的高温环境下的面内延迟(ro)的变化量增大的倾向。
式(6)所表示的二胺残基为具有两个苯环的芳香族二胺残基。对于成为式(6)所表示的二胺残基的来源的二胺化合物来说,可认为通过至少一个直接键合于苯环的氨基与二价连结基a位于间位,聚酰亚胺分子链所具有的自由度增加而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(6)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-o-、-ch2-、-c(ch3)2-、-co-、-so2-、-s-。
式(6)所表示的二胺残基例如可举出由以下的二胺化合物所衍生的二胺残基:3,3′-二氨基二苯基甲烷、3,3′-二氨基二苯基丙烷、3,3′-二氨基二苯基硫醚、3,3′-二氨基二苯基砜、3,3-二氨基二苯基醚、3,4′-二氨基二苯基醚、3,4′-二氨基二苯基甲烷、3,4′-二氨基二苯基丙烷、3,4′-二氨基二苯基硫醚、3,3′-二氨基二苯甲酮、(3,3′-双氨基)二苯基胺等。
式(7)所表示的二胺残基为具有三个苯环的芳香族二胺残基。对于成为式(7)所表示的二胺残基的来源的二胺化合物来说,可认为通过至少一个直接键合于苯环的氨基与二价的连结基a位于间位,聚酰亚胺分子链所具有的自由度增加而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,若使用式(7)所表示的二胺残基,则聚酰亚胺的热塑性提高。这里,连结基a优选-o-。
式(7)所表示的二胺残基例如可举出由以下的二胺化合物所衍生的二胺残基:1,4-双(3-氨基苯氧基)苯、3-[4-(4-氨基苯氧基)苯氧基]苯胺、3-[3-(4-氨基苯氧基)苯氧基]苯胺等。
式(8)所表示的二胺为具有三个苯环的芳香族二胺残基。对于式(8)所表示的二胺残基来说,可认为通过直接键合于一个苯环的两个二价的连结基a彼此位于间位,聚酰亚胺分子链所具有的自由度增加而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(8)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-o-。
式(8)所表示的二胺残基例如可举出由以下的二胺化合物所衍生的二胺残基:1,3-双(4-氨基苯氧基)苯(tpe-r)、1,3-双(3-氨基苯氧基)苯(apb)、4,4′-[2-甲基-(1,3-亚苯基)双氧基]双苯胺、4,4′-[4-甲基-(1,3-亚苯基)双氧基]双苯胺、4,4′-[5-甲基-(1,3-亚苯基)双氧基]双苯胺等。
式(9)所表示的二胺残基为具有四个苯环的芳香族二胺残基。对于成为式(9)所表示的二胺残基的来源的二胺化合物来说,可认为通过至少一个直接键合于苯环的氨基与二价的连结基a位于间位,而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(9)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-o-、-ch2-、-c(ch3)2-、-so2-、-co-、-conh-。
式(9)所表示的二胺残基可举出由以下的二胺化合物所衍生的二胺残基:双[4-(3-氨基苯氧基)苯基]甲烷、双[4-(3-氨基苯氧基)苯基]丙烷、双[4-(3-氨基苯氧基)苯基]醚、双[4-(3-氨基苯氧基)苯基]砜、双[4-(3-氨基苯氧基)]二苯甲酮、双[4,4′-(3-氨基苯氧基)]苯甲酰苯胺等。
式(10)所表示的二胺残基为具有四个苯环的芳香族二胺残基。对于式(10)所表示的二胺残基来说,可认为通过直接键合于至少一个苯环的两个二价的连结基a彼此位于间位,聚酰亚胺分子链所具有的自由度增加而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(10)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-o-。
式(10)所表示的二胺残基可举出由4-[3-[4-(4-氨基苯氧基)苯氧基]苯氧基]苯胺、4,4′-[氧基双(3,1-亚苯氧基)]双苯胺等二胺化合物所衍生的二胺残基。
式(11)所表示的二胺残基为具有四个苯环的芳香族二胺残基。关于式(11)所表示的二胺残基,可认为通过具有至少两个醚键而具有高的弯曲性,有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(11)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-c(ch3)2-、-o-、-so2-、-co-。
式(11)所表示的二胺残基例如可举出由以下的二胺化合物所衍生的二胺残基:2,2-双[4-(4-氨基苯氧基)苯基]丙烷(bapp)、双[4-(4-氨基苯氧基)苯基]醚(bape)、双[4-(4-氨基苯氧基)苯基]砜(baps)、双[4-(4-氨基苯氧基)苯基]酮(bapk)等。
式(12)所表示的二胺残基为具有四个苯环的芳香族二胺残基。对于式(12)所表示的二胺残基来说,可认为由于在二苯基骨架的两侧分别具有弯曲性高的二价的连结基a,因此有助于提高聚酰亚胺分子链的柔软性。因此,通过使用式(12)所表示的二胺残基,聚酰亚胺的热塑性提高。这里,连结基a优选-o-。
式(12)所表示的二胺残基例如可举出由双[4-(3-氨基苯氧基)]联苯、双[4-(4-氨基苯氧基)]联苯等二胺化合物所衍生的二胺残基。
在热塑性聚酰亚胺中,通过选定所述四羧酸残基及二胺残基的种类、或应用两种以上的四羧酸残基或二胺残基的情况下的各自的摩尔比,可控制热膨胀系数、拉伸模数、玻璃转移温度等。另外,在热塑性聚酰亚胺中具有多种聚酰亚胺的结构单元的情况下,能以嵌段的形式存在,也可无规地存在,优选的是无规地存在。
热塑性聚酰亚胺的重量平均分子量例如优选10,000~400,000的范围内,更优选50,000~350,000的范围内。若重量平均分子量小于10,000,则有膜的强度降低而容易脆化的倾向。另一方面,若重量平均分子量超过400,000,则有粘度过度地增加而在涂敷操作时容易产生膜厚度不均一、条纹等不良的倾向。
(非热塑性聚酰亚胺及热塑性聚酰亚胺的合成)
通常聚酰亚胺可通过以下方式制造:使四羧酸二酐与二胺化合物在溶剂中反应,生成聚酰胺酸后进行加热闭环。例如使四羧酸二酐与二胺化合物以大致等摩尔而溶解在有机溶剂中,在0℃~100℃的范围内的温度下搅拌30分钟~24小时而进行聚合反应,由此获得作为聚酰亚胺的前驱物的聚酰胺酸。反应时,以所生成的前驱物在有机溶剂中成为5重量%~30重量%的范围内、优选10重量%~20重量%的范围内的方式将反应成分溶解。聚合反应中所用的有机溶剂例如可举出:n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n,n-二乙基乙酰胺、n-甲基-2-吡咯啶酮(nmp)、2-丁酮、二甲基亚砜(dmso)、六甲基磷酰胺、n-甲基己内酰胺、硫酸二甲酯、环己酮、二噁烷、四氢呋喃、二乙二醇二甲醚、三乙二醇二甲醚、甲酚等。也可将这些溶剂并用两种以上,进而也可并用二甲苯、甲苯那样的芳香族烃。另外,这种有机溶剂的使用量并无特别限制,优选的是调整为通过聚合反应所得的聚酰胺酸溶液的浓度成为5重量%~30重量%左右那样的使用量而使用。
所合成的聚酰胺酸通常有利的是以反应溶剂溶液的形式使用,视需要可浓缩、稀释或替换成其他有机溶剂。另外,聚酰胺酸通常溶剂可溶性优异,因此可有利地使用。聚酰胺酸的溶液的粘度优选500cps~100,000cps的范围内。若偏离该范围,则利用涂布机等进行涂敷操作时容易产生厚度不均一、条纹等不良。使聚酰胺酸加以酰亚胺化的方法并无特别限制,例如适合采用在所述溶剂中在80℃~400℃的范围内的温度条件下加热1小时~24小时的热处理。
<覆铜层叠板>
本实施方式的覆铜层叠板具备绝缘层及位于该绝缘层的至少一个面上的铜箔等铜层,绝缘层只要使用本实施方式的聚酰亚胺膜而形成即可。另外,为了提高绝缘层与铜层的粘接性,绝缘层的与铜层接触的层为热塑性聚酰亚胺层。铜层是设置在绝缘层的单面或两面上。即,本实施方式的覆铜层叠板可为单面覆铜层叠板(单面ccl),也可为双面覆铜层叠板(两面ccl)。单面ccl的情况下,将层叠在绝缘层的单面上的铜层视为本发明的“第一铜层”。两面ccl的情况下,将层叠在绝缘层的单面上的铜层视为本发明的“第一铜层”,将层叠在绝缘层中与层叠有第一铜层的面为相反侧的面上的铜层视为本发明的“第二铜层”。本实施方式的覆铜层叠板是对铜层进行蚀刻等而进行布线电路加工,形成铜布线,用作fpc。
覆铜层叠板例如也可通过以下方式制备:准备含有本实施方式的聚酰亚胺膜而构成的树脂膜,对其溅镀金属而形成种子层后,例如通过镀铜而形成铜层。
另外,覆铜层叠板也可通过以下方式制备:准备含有本实施方式的聚酰亚胺膜而构成的树脂膜,利用热压接等方法对其层压铜箔。
进而,覆铜层叠板也可通过以下方式制备:在铜箔上浇注含有作为聚酰亚胺的前驱物的聚酰胺酸的涂布液,进行干燥而制成涂布膜后,进行热处理而酰亚胺化,形成聚酰亚胺层。
(第一铜层)
本实施方式的覆铜层叠板中,第一铜层所使用的铜箔(以下有时记作“第一铜箔”)并无特别限定,例如可为压延铜箔也可为电解铜箔。
第一铜箔的厚度以优选13μm以下,更优选6μm~12μm的范围内为宜。若第一铜箔的厚度超过13μm,则将覆铜层叠板(或fpc)弯折时对铜层(或铜布线)施加的弯曲应力增大,由此耐弯折性降低。另外,从生产稳定性及操作性的观点来看,第一铜箔的厚度的下限值优选的是设定为6μm。
另外,第一铜箔的拉伸模数例如优选10gpa~35gpa的范围内,更优选15gpa~25gpa的范围内。在本实施方式中使用压延铜箔作为第一铜箔的情况下,若通过热处理而进行退火,则柔软性容易提高。因此,若铜箔的拉伸模数小于所述下限值,则在长条的第一铜箔上形成绝缘层的工序中,因加热而导致第一铜箔自身的刚性降低。另一方面,若拉伸模数超过所述上限值,则将fpc弯折时对铜布线施加更大的弯曲应力,由此其耐弯折性降低。此外,压延铜箔有其拉伸模数根据在铜箔上形成绝缘层时的热处理条件、或形成绝缘层后的铜箔的退火处理等而变化的倾向。因此,本实施方式中,只要最终获得的覆铜层叠板中,第一铜箔的拉伸模数在所述范围内即可。
第一铜箔并无特别限定,可使用市售的压延铜箔。
(第二铜层)
第二铜层是层叠在绝缘层的与第一铜层为相反侧的面上。第二铜层所使用的铜箔(第二铜箔)并无特别限定,例如可为压延铜箔也可为电解铜箔。另外,第二铜箔也可使用市售的铜箔。此外,也可使用与第一铜箔相同的铜箔作为第二铜箔。
本实施方式的覆铜层叠板优选的是通过下述评价方法所得的10mm的电路基板尺寸(fpc尺寸)的布线图案的累计换算尺寸变化量相对于布线宽与布线间隔之和的比率在试片内的面内不均一性为±2%以下。所谓该不均一性的值为±2%以下,是指蚀刻前后的长度方向(md方向)的尺寸变化量及宽度方向(td方向)的尺寸变化量均为2%以下。在该不均一性的值超过±2%的情况下,在由覆铜层叠板加工而成的fpc中,成为引起布线间或布线与端子的接触不良的原因,导致电路基板的可靠性或良率降低。这里,一面参照图1~图7,一面对本实施方式中使用的覆铜层叠板的尺寸稳定性的评价方法加以说明。该评价方法包括以下的工序(1)~工序(6)。
(1)准备试片的工序:
该工序中,像图1所例示那样,将长条的覆铜层叠板100以既定的长度切断,由此准备试片10。此外,在以下的说明中,将长条的覆铜层叠板100的长度方向定义为md方向,将宽度方向定义为td方向(对于试片10来说也相同)。试片10优选的是以成为接近正方形的形状的方式而使覆铜层叠板100的宽度(td方向的长度)与切断间隔(md方向的长度)大致相等。虽省略图示,但覆铜层叠板100具有绝缘树脂层、及层叠在该绝缘树脂层的单侧或两侧的铜层。
成为本评价方法的对象的覆铜层叠板100可使用利用任意的方法所制备的覆铜层叠板。例如覆铜层叠板100可通过以下方式制备:准备树脂膜,对其溅镀金属而形成种子层后,通过镀敷而形成铜层。另外,覆铜层叠板100也可通过利用热压接等方法将树脂膜与铜箔层压而制备。进而,覆铜层叠板100也可通过在铜箔上涂布树脂溶液形成绝缘树脂层而制备。
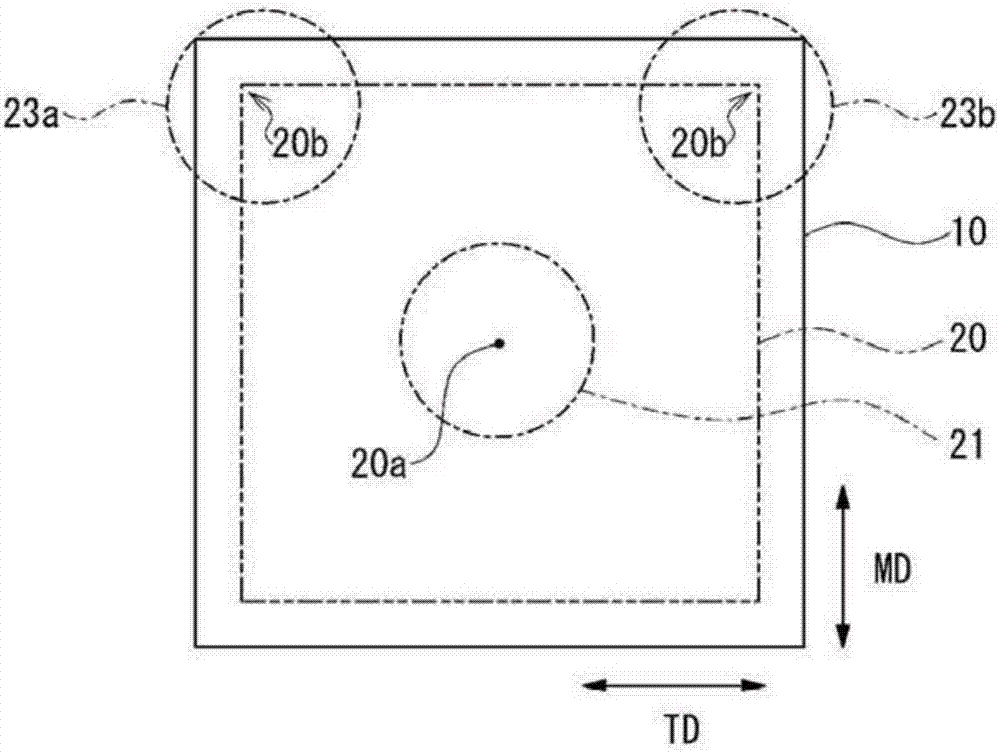
(2)在试片中形成多个标记的工序:
该工序中,像图2所示那样,首先在试片10中设想具有与md方向及td方向平行的边的假想正四边形20(以下有时简单地记作“正四边形20”)。该假想正四边形20的一边的长度可设定为与覆铜层叠板100的宽度(td方向的长度)相对应的长度。另外,关于假想正四边形20的面积,在采取多数个试片的情况下,加工成fpc的范围的极限包括在评价对象中,因此优选的是设定为可覆盖加工成fpc的范围的面积。因此,正四边形20的一边的长度优选的是设定为试片10的td方向的长度(覆铜层叠板100的宽度)的60%~90%的范围内,更优选的是设定为70%~80%的范围内。例如在覆铜层叠板100的宽度(td方向的长度)为250mm的情况下,假想正四边形20的一边的长度优选的是设定为150mm~225mm的范围内,更优选的是设定为175mm~200mm的范围内。
然后,像图2~图4所示那样,在假想正四边形20的包含中心20a的中心区域21、与逐一包含共有正四边形20的td方向一边的两个角部20b的两个角落区域23a、角落区域23b中,分别形成包含直线状的排列的多个标记。标记例如为贯穿试片10的圆孔30。多个孔30优选的是等间隔地形成。此外,作为标记的孔30例如可为三角形、长方形等多边形状。另外,标记只要可识别其位置,则不限于贯通孔,例如也可在试片10中形成槽、切口等,也可为利用油墨等印刷的花纹。
<中心区域>
假想正四边形20的中心20a成为用以测定试片10的伸缩的坐标的基准,因此该评价方法中,将包含该中心20a的中心区域21作为测定对象。中心区域21中,只要包含直线状的排列,则形成多个孔30的位置为任意,例如也可排列成t字形、l字形等,优选的是可从假想正四边形20的中心20a在md方向及td方向上均等地排列的十字型。即,像图3所示,优选的是沿着通过假想正四边形20的中心20a的十字形而在md方向及td方向上形成多个孔30,更优选的是以十字型的交叉部分与假想正四边形20的中心20a重叠的方式配置。该情况下,与中心20a重叠的孔30是作为构成md方向及td方向这两方向的排列的孔30而重复计数。
另外,中心区域21中,为了准确地评价包含试片10面内的尺寸变化不均一性的尺寸稳定性,以如下情况为宜:从正四边形20的中心20a起,在md方向及td方向上分别相对于正四边形20的一边长度而在至少12.5%以上、优选12.5%~32.5%的范围内、更优选12.5%~25%的范围内形成孔30。
<角落区域>
在图1所示那样的长条的覆铜层叠板100中,共有正四边形20的td方向一边的两个角部20b的周围为最易伸缩,尺寸变化容易变大的区域。因此,该评价方法中,将逐一包含共有正四边形20的td方向一边的两个角部20b的两个角落区域23a、角落区域23b两者作为测定对象。
角落区域23a、角落区域23b中,只要包含直线状的排列,则形成孔30的位置为任意,例如优选的是像图4所示那样,沿着假想正四边形20的夹持角部20b的两条边,在md方向及td方向上将多个孔30形成为l字形。该情况下,在角部20b重叠的孔30是作为构成md方向及td方向这两方向的排列的孔30而重复计数。此外,图4仅示出了一个角落区域23b,但对于另一角落区域23a来说也相同。
两个角落区域23a、角落区域23b中,为了可准确地评价包含试片10面内的尺寸变化不均一性的尺寸稳定性,以如下情况为宜:从正四边形20的td方向一边的两端(即正四边形20的角部20b)朝向md方向的中央侧,分别相对于md方向的一边长度而在至少12.5%以上、优选12.5%~32.5%的范围内、更优选12.5%~25%的范围内形成孔30。
另外,两个角落区域23a、角落区域23b中,为了可准确地评价包含试片10面内的尺寸变化不均一性的尺寸稳定性,以如下情况为宜:从正四边形20的td方向一边的两端(即正四边形20的角部20b)朝向td方向的中央侧,分别相对于td方向的一边长度而在至少12.5%以上、优选12.5%~32.5%的范围内、更优选12.5%~25%的范围内形成孔30。
另外,为了涵盖试片10的面内而准确地把握每个部位的尺寸变化,也可使中心区域21中排列成直线状的两端的孔30间的排列范围,与角落区域23a、角落区域23b中在相同方向上排列成直线状的两端的孔30间的排列范围重叠。
具体而言,也能以如下方式配置:在td方向上平行移动时,至少在中心区域21内排列在md方向上的多个孔30的两端的位置,与在两个角落区域23a、角落区域23b中分别排列在md方向上的多个孔30中最靠内侧(远离角部20b的一侧)的孔30的位置重叠(overlap)。
同样地,也能以如下方式配置:在md方向上平行移动时,至少在中心区域21内排列在td方向上的多个孔30中最接近角落区域23a、角落区域23b的孔30的位置,与在两个角落区域23a、角落区域23b内分别排列在td方向上的多个孔30中最靠内侧(远离角部20b的一侧)的孔30的位置重叠。
若考虑到如上配置,则中心区域21中将多个孔30排列成十字形最合理,另外,两个角落区域23a、角落区域23b中,将多个孔30排列成l字形最合理。
在试片10的假想正四边形20中,形成孔30的范围可通过孔30的大小、孔30的个数、孔30与孔30的间隔的长度而调节。
为了提高尺寸变化的检测精度,孔30的大小优选的是设定为孔30与孔30的间隔的长度的20%以下的范围内。
对于形成在所述中心区域21及两个角落区域23a、角落区域23b中的多个孔30来说,为了可准确地评价包含试片10面内的尺寸变化不均一性的尺寸稳定性,优选的是在md方向及td方向上分别包含至少11个以上的直线状的排列,更优选包含20个以上的直线状的排列。这里,若将孔30的个数设为n个,则后续工序(3)、工序(5)中成为测量对象的相邻的孔30与孔30的间隔数成为n-1处。关于相邻的孔30与孔30的间隔,例如在孔30的个数为10个的情况下成为9处,在孔30的个数为21个的情况下成为20处。该情况下,优选的是在md方向及td方向上,孔30的个数相同。
为了提高尺寸变化的检测精度,孔30与孔30之间的距离优选的是设定为2mm以上。
(3)第一测量工序:
该工序中,测定多个孔30的位置。而且,根据各孔30的位置的测定结果而算出邻接的孔30与孔30之间的距离l0。例如若孔30的个数为21个,则对邻接的孔30与孔30之间的20处的间隔求出距离l0。这里,像图5所示那样,邻接的孔30与孔30之间的距离l0是指从某个孔30的中心30a到邻接的孔30的中心30a的距离。
孔30的位置的测量并无特别限定,例如可通过根据试片10的图像来检测孔30的位置的方法而实施。
该工序的孔30的位置的测量可继所述工序(2)之后而实施,但优选的是在测量前设置调整试片10的状态(condition)的工序。试片10的状态调整的一例可举出调湿处理。调湿处理可通过将试片10在一定环境下静置一定时间(例如23℃、50rh%的环境下24小时)而进行。
(4)蚀刻工序:
该工序中,将试片10的铜层的一部分或全部蚀刻。为了评价切合实际的尺寸稳定性,蚀刻的内容优选的是依照由覆铜层叠板100所形成的fpc的布线图案而进行。在试片10是由双面覆铜层叠板所制备的情况下,也可对两侧的铜层进行蚀刻。此外,在实际的fpc加工中伴有热处理的情况下,也可在蚀刻后进行以任意温度将试片10加热的处理。
(5)第二测量工序:
该工序为在所述(4)的蚀刻后再次测定多个孔30的位置的工序。而且,像图5所示那样,根据各孔30的位置的测定结果而算出邻接的孔30与孔30之间的距离l1。该工序中的孔30的位置的测量可利用与所述工序(3)相同的方法来进行。
该工序的孔30的位置的测量可继所述工序(4)之后而实施,但优选的是与所述工序(3)同样地设置调整试片10的状态的工序。尤其在所述工序(3)中进行了状态调整的情况下,该工序中也优选的是在测量前以相同的条件实施状态调整。
(6)算出尺寸变化量的工序:
该工序中,像图5所示那样,在蚀刻前后对相同的两个孔30的间隔算出第一测量工序中所得的距离l0、与第二测量工序中所得的距离l1之差l1-l0。而且,对排列成同一直线状的孔30与孔30的间隔的2处以上、优选10处以上、更优选全部同样地算出差l1-l0。将该差l1-l0作为“尺寸变化量δ”。
(7)换算成布线规格的工序:
该工序中,将工序(6)中所得的尺寸变化量δ换算成由覆铜层叠板100所形成的fpc中的布线图案的规格,以相对于布线图案的布线宽与布线间隔之和的比率来表示所得的换算值。通过该工序,在将供试验的覆铜层叠板100实际加工成fpc的情况下,可容易地得知并表现出覆铜层叠板100的尺寸变化对fpc的布线图案造成的影响。
该工序中,首先将尺寸变化量δ换算成由覆铜层叠板100所形成的预定的fpc中的l/s布线图案的布线宽/布线间隔的规格,将换算所得的尺寸变化量累计而求出累计换算尺寸变化量。例如在相对于蚀刻前的两个孔30之间的距离l0,预定形成的fpc中的布线图案的布线宽与布线间隔分别为距离l0的1/y的情况下,根据下式将尺寸变化量δ换算成缩减(downsizing)为2×(1/y)的规格时的值,求出2×(1/y)的规格的累计换算尺寸变化量。
累计换算尺寸变化量=[∑i=11(2×δi/y)]
所述式中,记号∑i=11表示1至i的总和。另外,尺寸变化量δ表示蚀刻后的第n个孔30与第n-1个孔30的距离l1减去蚀刻前的第n个孔30与第n-1个孔30的距离l0所得的值(这里,n为2以上的整数)。例如,δ1为第1个间隔的长度(相邻的两个孔30间的距离)的尺寸变化量,δi为第i个(i是指正整数)间隔的长度的尺寸变化量。
然后,由累计换算尺寸变化量根据下式求出布线的位移比率。该布线的位移比率是以相对于预定形成的l/s布线图案的布线宽(lmm)与布线间隔(smm)之和的比率来表示累计换算尺寸变化量。
布线的位移比率(%)=
{[∑i=11(2×δi/y)]/[l+s]}×100
将像以上那样所算出的fpc中的md及td的布线的位移比率绘制在图表上,由此获得与fpc尺寸相对应的近似直线(此外,省略图表的图示)。这里,所谓“fpc尺寸”,是指fpc中形成的多条布线中距离最远的两端的布线间的距离。图表的斜率的大小是指布线位移的大小,图表的斜率的不均一性的大小是指布线位移的面内不均一性的大小。
通过该工序,在将供试验的覆铜层叠板100实际加工成电路的情况下,可容易地得知并表现出覆铜层叠板100的尺寸变化对fpc的布线图案造成的影响。另外,通过制作近似直线的图表,可与fpc尺寸相对应而看到并表现出由作为供试体的覆铜层叠板100所制作的布线的位移的大小或面内的不均一性。
此外,也可将所述工序(6)中所得的尺寸变化量δ累计后,将累计尺寸变化量换算成由覆铜层叠板100所形成的预定的fpc中的l/s布线图案的布线宽/布线间隔的规格,求出累计换算尺寸变化量。例如将各间隔的尺寸变化量δ累计,获得累计尺寸变化量∑。该累计尺寸变化量∑可通过下式而算出。
∑=δ1+δ2+δ3+···+δi=∑i=11δi
累计尺寸变化量∑可对覆铜层叠板100的md方向、td方向的任一方向、优选两个方向求出。根据累计尺寸变化量∑的大小,可评价覆铜层叠板100的md方向、td方向的尺寸稳定性。另外,根据累计尺寸变化量∑的实测值而获得规格增大(scaleup)的近似直线。
像以上那样,根据该评价方法,可通过工序(1)~工序(7)而高精度地评价包含面内不均一性的覆铜层叠板100的尺寸变化。另外,即便在从覆铜层叠板100采取多数个试片的情况下,也可对加工成fpc的各区域分别评价尺寸稳定性。
<fpc>
本实施方式的覆铜层叠板主要作为fpc材料而有用。即,利用常法将本实施方式的覆铜层叠板的铜层加工成图案状而形成布线层,由此可制造作为本发明的一实施方式的fpc。
[实施例]
以下示出实施例对本发明的特征进行更具体说明。然而,本发明的范围不限定于实施例。此外,以下的实施例中,只要无特别说明,则各种测定、评价是利用下述方法。
[粘度的测定]
关于粘度的测定,使用e型粘度计(博勒飞(brookfield)公司制造,商品名:dv-ii+pro)测定25℃下的粘度。以扭矩(torque)成为10%~90%的方式设定转速,开始测定起经过2分钟后,读取粘度稳定时的值。
[重量平均分子量的测定]
重量平均分子量是利用凝胶渗透色谱仪(东曹(tosoh)股份有限公司制造,商品名:hlc-8220gpc)进行测定。使用聚苯乙烯作为标准物质,展开溶剂使用n,n-二甲基乙酰胺。
[玻璃转移温度(tg)的测定]
关于玻璃转移温度,使用动态粘弹性测定装置(dma:ubm公司制造,商品名:e4000f),从30℃到400℃以升温速度4℃/分钟、频率11hz对尺寸为5mm×20mm的聚酰亚胺膜进行测定,将弹性模数变化(tanδ)达到最大的温度作为玻璃转移温度。此外,将使用dma所测定的30℃下的储存模数为1.0×109pa以上、360℃下的储存模数小于1.0×108pa者视为“热塑性”,将30℃下的储存模数为1.0×109pa以上、360℃下的储存模数显示出1.0×108pa以上者视为“非热塑性”。
[热膨胀系数(cte)的测定]
使用热机械分析仪(布鲁克(bruker)公司制造,商品名:4000sa),对尺寸为3mm×20mm的聚酰亚胺膜一面施加5.0g的负重,一面以一定的升温速度使其从30℃升温到265℃,进而在该温度下保持10分钟后,以5℃/分钟的速度冷却,求出250℃至100℃的平均热膨胀系数(热膨胀系数)。
[铜箔的表面粗度的测定]
关于铜箔的表面粗度,使用原子力显微镜(atomicforcemicroscope,afm)(布鲁克axs(brukeraxs)公司制造,商品名:维度图标(dimensionicon)型spm)、探针(布鲁克axs(brukeraxs)公司制造,商品名:tespa(nchv),前端曲率半径10nm,弹簧常数42n/m),以轻敲模式(tappingmode)对铜箔表面的80μm×80μm的范围进行测定,求出十点平均粗糙度(rz)。
[剥离强度的测定]
1)单面覆铜层叠板的浇注侧(树脂涂敷侧)
将单面覆铜层叠板(铜箔/树脂层)的铜箔以宽度1.0mm进行电路加工后,以宽度:8cm×长度:4cm切断,制备测定样本1。关于测定样本1的浇注侧的剥离强度,使用拉伸测试机(tensilontester)(东洋精机制作所制造,商品名:万能试验机(strograph)ve-1d),利用双面胶带将测定样本1的树脂层侧固定在铝板上,将铜箔朝90°方向以50mm/分钟的速度剥离,求出将铜箔从树脂层剥离10mm时的中央强度。将该值作为剥离强度1a。
2)双面覆铜层叠板的浇注侧(树脂涂敷侧)
将双面覆铜层叠板(铜箔/树脂层/铜箔)的热压接侧与浇注侧两面的铜箔以宽度0.8mm进行电路加工(以两面的铜箔成为相同位置的方式进行布线加工)后,以宽度:8cm×长度:4em切断,制备测定样本2。关于测定样本2的浇注侧的剥离强度,使用拉伸测试机(tensilontester)(东洋精机制作所制造,商品名:万能试验机(strograph)ve-1d),利用双面胶带将测定样本2的热压接侧的铜箔面固定在铝板上,将铜箔朝90°方向以50mm/分钟的速度剥离,求出将树脂涂敷侧的铜箔从树脂层剥离10mm时的中央值强度。将该值作为剥离强度2a。
[面内延迟(ro)的测定]
关于面内延迟(ro),使用双折射率计(光子晶格(photonic-lattice)公司制造,商品名:宽范围(widerange)双折射评价系统wpa-100,测定区域:md:140mm×td:100mm),求出既定样本的面内方向的延迟。此外,入射角为0°,测定波长为543nm。
[拉伸模数的测定]
关于铜箔的拉伸模数,应用使用真空烘箱进行了与覆铜层叠板的处理工序同等的热处理的铜箔,使用东洋精机制作所股份有限公司制造的万能试验机(strograph)r-1,在温度23℃、相对湿度50%的环境下测定拉伸模数的值。
实施例及比较例中所用的简称表示以下的化合物。
ntcda:2,3,6,7-萘四羧酸二酐
bpda:3,3′,4,4′-联苯四羧酸二酐
pmda:均苯四甲酸二酐
m-tb:2,2′-二甲基-4,4′-二氨基联苯
m-eob:2,2′-二乙氧基-4,4′-二氨基联苯
tpe-r:1,3-双(4-氨基苯氧基)苯
dape:4,4′-二氨基二苯基醚
bapp:2,2-双[4-(4-氨基苯氧基)苯基]丙烷
dmac:n,n-二甲基乙酰胺
(合成例1)
在氮气流下,在反应槽中投入1146.4重量份的m-tb(5.4摩尔份)及175.4重量份的tpe-r(0.6摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加706.1重量份的bpda(2.4摩尔份)及965.4重量份的ntcda(3.6摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液1。聚酰胺酸溶液1的溶液粘度为41,100cps。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液1均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜1(联苯四基及亚联苯基:65摩尔%,非热塑性,tg:400℃以上,cte:7.7ppm/k)。
(合成例2)
在氮气流下,在反应槽中投入743.0重量份的m-tb(3.5摩尔份)及672.4重量份的tpe-r(2.3摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加353.1重量份的bpda(1.2摩尔份)及1233.6重量份的ntcda(4.6摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液2。聚酰胺酸溶液2的溶液粘度为41,900cps。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液2均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜2(联苯四基及亚联苯基:41摩尔%,非热塑性,tg:391℃,cte:19.1ppm/k)。
(合成例3)
在氮气流下,在反应槽中投入1040.2重量份的m-tb(4.9摩尔份)及350.8重量份的tpe-r(1.2摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加529.6重量份的bpda(1.8摩尔份)、643.6重量份的ntcda(2.4摩尔份)及392.6重量份的pmda(1.8摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液3。聚酰胺酸溶液3的溶液粘度为32,500cps。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液3均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜3(联苯四基及亚联苯基:55摩尔%,非热塑性,tg:377℃,cte:14.8ppm/k)。
(合成例4)
在氮气流下,在反应槽中投入552.0重量份的m-tb(2.6摩尔份)及760.9重量份的dape(3.8摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加1716.3重量份的ntcda(6.4摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液4。聚酰胺酸溶液4的溶液粘度为42,300cps。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液4均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜4(亚联苯基:20摩尔%,非热塑性,tg:400℃以上,cte:32.1ppm/k)。
(合成例5)
在氮气流下,在反应槽中投入898.7重量份的m-eob(3.3摩尔份)及660.8重量份的dape(3.3摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加1439.6重量份的pmda(6.6摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液5。聚酰胺酸溶液5的溶液粘度为31,700cps。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液5均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜5(亚联苯基:25摩尔%,非热塑性,tg:376℃,cte:33.5ppm/k)。
(合成例6)
在氮气流下,在反应槽中投入63.7重量份的m-tb(0.3摩尔份)及1490.9重量份的tpe-r(5.1摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加1118.0重量份的bpda(3.8摩尔份)及349.0重量份的pmda(1.6摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液6。聚酰胺酸溶液6的溶液粘度为6,700cps,重量平均分子量为163,400。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液6均匀涂布在不锈钢制的支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜6(联苯四基及亚联苯基:38摩尔%,热塑性,tg:242℃,30℃的储存模数:4.3×109pa,360℃的储存模数:1.4×107pa)。
(合成例7)
在氮气流下,在反应槽中投入743.0重量份的m-tb(3.5摩尔份)及672.4重量份的tpe-r(2.3摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加1206.3重量份的bpda(4.1摩尔份)及370.8重量份的pmda(1.7摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液7。聚酰胺酸溶液7的溶液粘度为7,200cps,重量平均分子量为112,000。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液7均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜7(联苯四基及亚联苯基:66摩尔%,热塑性,tg:266℃,30℃的储存模数:4.3×109pa,360℃的储存模数:7.1×107pa)。
(合成例8)
在氮气流下,在反应槽中投入233.5重量份的m-tb(1.1摩尔份)及1344.7重量份的tpe-r(4.6摩尔份)以及聚合后的固体成分浓度成为15重量%的量的dmac,在室温下搅拌而加以溶解。然后,添加676.7重量份的bpda(2.3摩尔份)及741.6重量份的pmda(3.4摩尔份)后,在室温下继续搅拌3小时而进行聚合反应,获得聚酰胺酸溶液8。聚酰胺酸溶液8的溶液粘度为7,400eps,重量平均分子量为163,400。
然后,以硬化后的厚度成为约25μm的方式将聚酰胺酸溶液8均匀涂布在不锈钢制支撑基材上后,在120℃下加热干燥而除去溶剂。进而,从120℃到360℃在30分钟以内进行阶段性的热处理,完成酰亚胺化,制备聚酰亚胺膜8(联苯四基及亚联苯基为30摩尔%,热塑性,tg:279℃,30℃的储存模数:4.1×109pa,360℃的储存模数:7.9×107pa)。
[实施例1-1]
在环形带状的不锈钢制支撑基材上,使用多歧管(multimanifold)式的三层共挤出多层模头,以聚酰胺酸溶液7/聚酰胺酸溶液1/聚酰胺酸溶液7的顺序的三层结构连续地挤出涂布,在130℃下加热干燥3分钟而除去溶剂。然后,从130℃到360℃进行阶段性的热处理,完成酰亚胺化,制备热塑性聚酰亚胺层/非热塑性聚酰亚胺层/热塑性聚酰亚胺层的厚度分别为2.5μm/20μm/2.5μm的聚酰亚胺膜1a′。利用刀锋法将支撑基材上的聚酰亚胺膜1a′剥离,制备宽度方向的长度为1100mm的长条状聚酰亚胺膜1a。
<面内延迟(ro)评价用样本的制备>
将长条状聚酰亚胺膜1a的td方向的左右两个端部(左侧(left)及右侧(right))以及中央部(center)分别以a4尺寸(td:210mm×md:297mm)切断,制备样本l1(left)、样本r1(right)及样本c1(center)。
<面内延迟(ro)的评价>
对样本l1、样本r1及样本c1各自分别测定面内延迟(ro)。将各样本的测定值的最大值作为“面内延迟(ro)”,将面内延迟(ro)的测定值中的最大值与最小值之差作为“宽度方向(td方向)的面内延迟(ro)的不均一性(δro)”。另外,“在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量”是设定为样本c1的加压前及加压后的各面内延迟(ro)的测定值中的最大值之差。
此外,各样本中的测定区域如下。
样本l1:td方向的左侧端部区域及md方向的中央区域
样本r1:td方向的右侧端部区域及md方向的中央区域
样本c1:td方向及md方向的中央区域
长条状聚酰亚胺膜1a的评价结果如下。
cte:17ppm/k
面内延迟(ro):11nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):1nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:3nm
[实施例1-2]
除了将宽度方向(td方向)的长度设为540mm以外,与实施例1-1同样地制备长条状聚酰亚胺膜1b。
长条状聚酰亚胺膜1b的评价结果如下。
cte:17ppm/k
面内延迟(ro):11nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):1nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:3nm
[实施例1-3]
在长条状的铜箔1(压延铜箔,jx金属股份有限公司制造,商品名:ghy5-93f-ha-v2箔,厚度:12μm,热处理后的拉伸模数:18gpa,宽度方向的长度:540mm)的表面上,以硬化后的厚度成为2.5μm的方式均匀涂布聚酰胺酸溶液7后,在120℃下加热干燥1分钟而除去溶剂。在其上以硬化后的厚度成为20μm的方式均匀涂布聚酰胺酸溶液1后,在120℃下加热干燥3分钟而除去溶剂。进而,在其上以硬化后的厚度成为2.5μm的方式均匀涂布聚酰胺酸7后,在120℃下加热干燥1分钟而除去溶剂。然后,从130℃到360℃进行阶段性的热处理,完成酰亚胺化后,制备单面覆铜层叠板1a。
对单面覆铜层叠板1a的铜箔进行蚀刻除去而制备的长条状聚酰亚胺膜的评价结果如下。
cte:17ppm/k
面内延迟(ro):11nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):1nm
在所述单面覆铜层叠板1a的聚酰亚胺层侧重合铜箔1,在温度360℃、压力340mpa/m2的条件下热压接15分钟,制备双面覆铜层叠板1a。对双面覆铜层叠板1a的铜箔进行蚀刻除去而制备的长条状聚酰亚胺膜的面内延迟(ro)为8nm。
对所制备的双面覆铜层叠板1a的中央部进行切割加工,准备长条状的覆铜层叠板1a′(端宽:250mm)作为尺寸稳定性的评价用样本的材料。
<尺寸稳定性的评价用样本的制备>
将所述覆铜层叠板1a′在md方向上以长度250mm切断,制成md:250mm×td:250mm。像图6所示那样,在切断后的覆铜层叠板的md:200mm×td:200mm的范围内设想假想正四边形。在该假想正四边形的逐一包含共有td方向一边的两个角部的左右两个角落区域(left及right)以及包含假想正四边形的中心的中央区域(center)中,分别在md及td方向上以2.5mm的间隔连续地进行21个开孔加工,制备评价用样本。此外,开孔加工使用直径0.105mm的钻头(drill)。
<尺寸稳定性的评价>
使用非接触计算机数控(computernumericalcontrol,cnc)图像测定机(三丰(mitutoyo)公司制造,商品名:快速视像(quickvision)qv-x404pil-c),对将评价用样本的两面的铜箔层全部蚀刻而除去前后的各孔的位置进行测定。根据测定值算出蚀刻前后的相邻两孔间距离的尺寸变化量及累计尺寸变化量。
准备长条状的覆铜层叠板1a′,像图7所示那样制备评价用样本1、评价用样本2。对评价用样本1、评价用样本2分别测定center、left及right的蚀刻前后的各孔的位置。根据测定值算出蚀刻前后的相邻两孔间的距离的尺寸变化量及这些的合计(20处)的累计尺寸变化量。
根据覆铜层叠板1a′的评价结果,将md的累计尺寸变化量及不均一性示于表1中。此外,表1中,以left、center、right的累计尺寸变化率以及将累计尺寸变化量换算成设想fpc尺寸10mm的累计换算尺寸变化量来表示,也示出left、center、right的整个范围的不均一性。此外,“累计尺寸变化率”是指累计尺寸变化量相对于蚀刻前的两孔间距离的合计值的比率(%)。另外,表中的“范围”的数值是指left、center、right的整个范围的中央值±上下范围(表2、表3中相同)。
[表1]
根据该结果,确认到可对将覆铜层叠板1a′作为材料而形成的电路布线基板(l/s=0.025mm/0.0025mm)评价布线的位移比率及试片面内的尺寸变化率的不均一性,并且可确认实施例1-3的双面覆铜层叠板1a的各fpc尺寸时的布线的位移比率的不均一性小。另外可确认,为了实现仅控制聚酰亚胺膜的cte的情况下无法达成的覆铜层叠板的高的尺寸稳定精度,重要的是控制面内延迟。
[实施例1-4]
在实施例1-2中制备的长条状聚酰亚胺膜1b的两面上重合铜箔1,以温度360℃、压力340mpa/m2的条件热压接15分钟,制备双面覆铜层叠板1b,与实施例1-3同样地准备长条状的覆铜层叠板1b′(端宽:250mm),进行尺寸稳定性的评价。将其结果示于表2中。
[表2]
[比较例1-1]
准备长条状聚酰亚胺膜1c(厚度:25μm,钟渊(kaneka)公司制造,商品名:皮克斯奥(pixeo))。
长条状聚酰亚胺膜1c的评价结果如下。
cte:17ppm/k
面内延迟(ro):200nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):80nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:30nm
[比较例1-2]
在长条状聚酰亚胺膜1c的两面上重合铜箔1,以温度360℃、压力340mpa/m2的条件热压接15分钟,制备双面覆铜层叠板1c,与实施例1-3同样地制备长条状的覆铜层叠板1c′(端宽:250mm),进行尺寸稳定性的评价。将其结果示于表3中。
[表3]
[实施例2-1]
除了聚酰胺酸溶液8/聚酰胺酸溶液1/聚酰胺酸溶液8的顺序的三层结构以外,与实施例1-1同样地制备宽度方向的长度为1100mm的长条状聚酰亚胺膜2。
长条状聚酰亚胺膜2的评价结果如下。
cte:17ppm/k
面内延迟(ro):11nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):1nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:5nm
[实施例3-1]
除了聚酰胺酸溶液6/聚酰胺酸溶液2/聚酰胺酸溶液6的顺序的三层结构以外,与实施例1-1同样地制备宽度方向的长度为1100mm的长条状聚酰亚胺膜3。
长条状聚酰亚胺膜3的评价结果如下。
cte:17ppm/k
面内延迟(ro):17nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):3nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:7nm
[实施例4-1]
除了聚酰胺酸溶液8/聚酰胺酸溶液3/聚酰胺酸溶液8的顺序的三层结构以外,与实施例1-1同样地制备宽度方向的长度为1100mm的长条状聚酰亚胺膜4。
长条状聚酰亚胺膜4的评价结果如下。
cte:17ppm/k
面内延迟(ro):15nm
宽度方向(td方向)的面内延迟(ro)的不均一性(δro):2nm
在温度360℃的环境下以压力340mpa/m2、保持时间15分钟加压前后的面内延迟(ro)的变化量:10nm
以上,以例示为目的对本发明的实施方式进行了详细说明,但本发明不受所述实施方式的限制,可进行各种变形。
本申请案主张基于2016年4月27日提出申请的日本专利申请案2016-89514号的优先权,将该申请案的所有内容引用至本文中。
- 还没有人留言评论。精彩留言会获得点赞!