一种提高乳酸菌素plnJ的产量的基因工程方法与流程
本发明属于生物技术领域,具体涉及一种提高乳酸菌素plnj的产量的基因工程方法。
背景技术:
抗生素作为畜禽抑菌药物和促生长剂在畜牧业生产中得到了广泛应用。然而长期使用抗生素,不但影响动物疾病的防治效果;还会产生抗药性和残留,造成严重的畜产品污染,有的甚至会诱发人和动物产生癌变和畸变,危及人类的健康与安全。因此,2006年1月1日起,欧盟全面禁止食品动物使用抗生素促生长饲料添加剂。全面禁止抗生素作为食品动物促生长饲料添加剂,越来越来引起全球的普遍重视。然而,欧盟由于全面禁止抗生素饲料添加剂也给动物生产带来一些问题,如肠道细菌疾病的增加。为了扭转动物健康的下降,只能大量使用治疗性抗生素来治疗。同时,由于肠道细菌性疾病的增加也增加了动物性食品被肠道细胞污染的机会如沙门氏菌污染。为此,研制一种能改善畜禽健康,提高生产性能且安全无毒的能替代抗生素的产品已成为动物营养研究的一个热点。
乳酸菌素是由某些乳酸菌细菌在代谢过程中通过核糖体合成机制产生的具有抑菌活性的多肽或前体多肽。许多研究表明,发酵过程中如菌株、培养条件(最适生长温度、培养时间、最适ph值、装液量等)及培养基成分(例如碳源、氮源、磷酸盐等)对细菌素产量具有重要的影响。乳酸菌素plnj是乳酸菌的分泌的细菌素中的重要一种。由于野生型乳酸菌其分泌乳酸菌素plnj的产量同样受到多种因素的影响,因此,探索一种有效的提高乳酸菌素plnj产量的方法对于规模化生产乳酸菌素plnj具有重要的意义。
基因工程方法主要是通过构建重组质粒,转染工程菌,利用工程菌高效表达的特点,将外来的蛋白进行大量的生产。因此,通过研究,我们建立了能够有效提高乳酸菌素plnj的基因工程方法,体外表达了乳酸菌素plnj,并验证了该乳酸菌素plnj仍然具有较高的广谱抑菌功能。
技术实现要素:
本发明的目的在于提供一种提高乳酸菌素plnj的产量的基因工程方法。
为实现上述目的,本发明采用如下技术方案:
引物的设计与合成:参考genebank乳酸菌(genebank:x94434)的plnj基因的核苷酸序列,采用primerpremier6设计一对特异性引物,plnj上游引物f:5’-gcggatccatgactgtgaacaaaatgat-3’,plnj下游引物r:5’-atgtcgacttaacgacggattgctctgc-3’,将设计好的引物序列送上海生工生物工程股份有限公司合成。
一种提高乳酸菌素plnj的产量的基因工程方法,包括如下步骤:
(1)设计一对plnj特异性引物,plnj上游引物f:5’-gcggatccatgactgtgaacaaaatgat-3’,plnj下游引物r:5’-atgtcgacttaacgacggattgctctgc-3’;
(2)以plnj基因的上游引物f和下游引物r作为扩增引物,乳酸菌dna为模板,对plnj基因进行pcr扩增,pcr扩增产物用琼脂糖凝胶电泳鉴定;
(3)琼脂糖凝胶电泳鉴定pcr产物后,切下目的条带进行纯化;将纯化后的plnj产物与质粒pet-32a(+)进行双酶切;取酶切的pcr目的片段和载体片段进行连接反应;将连接后的重组质粒,转化至bl21感受态细胞,获得能够在体外诱导表达的重组蛋白。
本发明的优点在于:
(1)利用基因工程技术体外重组表达乳酸菌素plnj,该技术方便简单,通过扩大培养工程菌就可以大规模生产乳酸菌素plnj。
(2)体外重组表达乳酸菌素plnj仍然具有广谱抑菌功能,且具有安全、无毒、无副作用、无残留等优点,在动物生产上可以作为饲料添加剂。
附图说明
图1pcr扩增结果。其中m:dna分子质量标准;1:plnj基因扩增的产物。
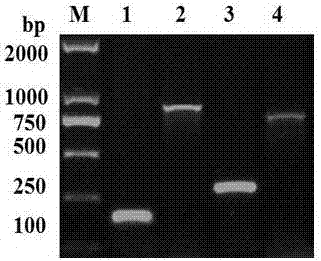
图2pcr鉴定结果。其中m:dnamarker;1:plnj的pcr扩增产物;2:t7引物的pcr扩增产物:3:t7上游引物和plnj下游引物的pcr扩增结果;4:t7下游引物和plnj上游引物pcr产物。
图3酶切鉴定结果。m1、m2:dnamarker;1:未酶切的pet-32a(+)-plnj重组质粒;2:pet-32a(+)-plnj重组质粒的bamhi单酶切;3:pet-32a(+)-plnj重组质粒重组的sali单酶切;4:pet-32a(+)-plnj重组质粒的bamhi+sali双酶切。
图4iptg最佳诱导温度的优化结果。其中m:蛋白质分子质量标准;1:空白组诱导组;2~6:分别为16℃,25℃,31℃,37℃,43℃梯度温度下诱导的pet-32a(+)-plnj重组质粒菌组。
图5iptg最佳诱导时间的优化结果。其中m:蛋白质分子质量标准;1:空白组诱导组;2~6:分别为诱导后0h,4h,8h,12h,16h梯度下诱导的pet-32a(+)-plnj重组质粒菌组。
图6iptg最佳诱导浓度的优化结果。其中m:蛋白质分子质量标准;1:空白组诱导组;2~9:分别为iptg诱导浓度为0mmol/l,0.2mmol/l,0.4mmol/l,0.6mmol/l,0.8mmol/l,1.0mmol/l,1.2mmol/l,1.5mmol/l梯度下诱导的pet-32a(+)-plnj重组质粒菌组。
图7重组蛋白的ni-nta亲和层析纯化结果。其中m:蛋白质分子质量标准;1:纯化前蛋白溶液2~9:分别为40mm,80mm,120mm,160mm,200mm,240mm,280mm和320mm等咪唑洗脱液的纯化结果。
图8western-blot鉴定结果。其中11:pet-32a(+)-plnj组;2:pet-32a(+)组;3:空白菌组。
具体实施方式
1材料与方法
1.1材料
1.1.1菌株及表达载体
菌株:由本福建农林大学动物科学学院组织胚胎学实验室分离并保存的乳酸菌fq株;表达的宿主菌:e.colidh5α感受态细胞,e.colibl21(de3)感受态细胞(购自北京全式金生物公司);表达载体:pet-32a(+)(购自promega北京生物技术有限公司)1。
1.1.2主要试剂
细菌基因组dna提取试剂盒(dp302)、质粒小提中量试剂盒(dp106-02)(购自北京天根生化科技有限公司);琼脂糖凝胶dna快速回收试剂盒(cw2302)(购自北京康为世纪生物技术公司);t4连接酶(购自promega公司);bamhiquickcut限制酶、saliquickcut限制酶、(购自大连宝生物工程有限公司);transtaqtmdnapolymerasehighfidelity(hifi)(ap131-11)、proteinisotmni-ntaresin蛋白纯化柱(dp101-02)、bca蛋白浓度测定试剂盒、辣根过氧化物酶(hrp)标记的羊抗鼠及抗his标签蛋白一抗(购自北京全式金公司);blueplusivproteinmarker(10kda~180kda);supersignalwestpico化学发光底物(购自赛默飞世尔科技公司);sds-page凝胶制备试剂盒(ar0138)、异丙基-β-d-硫代半乳糖苷(iptg);其余常规试剂与药品均购自国药公司。
1.2试验方法
1.2.1引物的设计与合成
参考genebank乳酸菌(genebank:x94434)的plnj基因的核苷酸序列,采用primerpremier6设计一对特异性引物,plnj上游引物f:5’-gcggatccatgactgtgaacaaaatgat-3’,plnj下游引物r:5’-atgtcgacttaacgacggattgctctgc-3’将设计好的引物序列送上海生工生物工程股份有限公司合成。
1.2.2plnj基因的扩增
1.2.2.1乳酸菌总dna的提取
按照细菌基因组dna提取试剂盒说明书提取乳酸菌的dna。
1.2.2.2pcr扩增
以plnj基因的上游引物f和下游引物r作为扩增引物,乳酸菌dna为模板,对plnj基因进行pcr扩增,pcr扩增产物用琼脂糖凝胶电泳鉴定,具体pcr反应体系和反应条件(见下表1)。
表1pcr扩增体系表和反应条件
1.2.3重组表达质粒的构建与鉴定
1.2.3.1pcr扩增产物的纯化
琼脂糖凝胶电泳鉴定pcr产物后,切下目的条带进行纯化,详细步骤参照常规方法进行。
1.2.3.2质粒pet-32a(+)的提取
将实验室冻存的质粒pet-32a(+)进行复苏,按1%的接种量接种到10ml的含有氨苄的lb肉汤中,置于37℃的恒温摇床中,200rpm培养过夜,使用质粒小提中量试剂盒提取质粒。
1.2.3.2目的片段与表达载体的双酶切
将纯化后的plnj产物与提取得到的质粒pet-32a(+)进行双酶切。质粒pet-32a(+)采用100ul的双酶切体系,纯化后的plnj采用60ul的双酶切体系。双酶切反应结束后,再次胶回收纯化酶切的目的片段和载体片段以备进行连接反应。
1.2.3.3连接反应
取再次胶回收纯化的酶切的pcr目的片段plnj和载体片段,进行琼脂糖凝胶电泳估算二者浓度,接着以目的片段与载体摩尔比例为7:1进行连接反应,摩尔与体积计算方法见t4连接酶说明书(promega公司),连接体系与反应条件(详见下表2):
表2连接反应体系及条件
1.2.3.4重组质粒转化
将连接后的重组质粒,转化至dh5α感受态细胞进行扩增,操作步骤如下:
(1)将50μl感受态细胞dh5α加入到连接产物中,在冰上放置25min;
(2)冰浴完成之后,42℃热激45s,并立即置于冰上放置2min;
(3)加入250μl平衡至室温的lb肉汤(不加amp+),200r/min,37℃振荡孵育培养1h;
(4)取200μl菌液均匀涂于含有lb的固体培养板(加amp+),倒置于37℃恒温培养箱中,过夜培养(为得到较多的克隆,4000r/min离心1min,弃掉部分上清,保留100~150μl,轻轻弹匀悬浮菌体,取全部菌液涂板)。
1.2.3.5pcr鉴定阳性重组子及重组质粒提取
挑选白色的单克隆菌落至10μl的无菌水中,吹打混匀,以此混合液为模板进行pcr鉴定阳性重组质粒或以载体上的t7通用引物(表3)进行扩增。同时将同一菌落接种于液体lb培养基(含amp),200r/min,37℃振荡培养过夜。利用质粒小提中量试剂盒提取pcr阳性的质粒dna。
表3t7引物序列表
1.2.3.6重组表达质粒的酶切鉴定及序列测定
提取pcr鉴定阳性的重组质粒进行双酶切鉴定,具体的酶切体系和条件参照常规方法。琼脂糖凝胶电泳鉴定pcr及酶切反应产物,鉴定结果符合预期的重组质粒送往上海生物工程股份有限公司测序并将测序结果正确的重组表达质粒命名为pet-32a(+)-plnj。
1.2.4乳酸菌素plnj基因序列的分析
利用ncbi网站核甘酸序列比对分析测序结果;同时运用mega生物软件进行基因同源性分析及推导的氨基酸序列分析。
1.2.5重组质粒pet-32a(+)-plnj在e.colibl21(de3)中的诱导表达
将重组质粒pet-32a(+)-plnj转化至e.colibl21(de3)感受态细胞中,方法同1.2.3.4所述,于平板中挑取白色单克隆菌落接种于1ml的液体lb培养基(含amp),置200r/min,37℃的摇床中培养至浓度约为d600nm=1.0,然后扩大培养,以1:100的比例接种于lb液体培养基(含amp)中,200r/min,37℃的摇床中培养,至浓度约为d600nm=1.0,分成两份,一份加入iptg至终浓度为1mmol/l诱导蛋白表达,另一份不加入iptg,继续置于摇床中培养12h,同时设pet-32a(+)空载体菌诱导组和空白诱导组。取500μl的菌群悬液,8000r/min,4℃离心5min,收集菌群,用100μlpbs缓冲液重悬,按照稀释比例加入6×sds-page上样缓冲液,振荡混匀后于沸水中煮沸5min,冰上冷却5min,于4℃,12000r/min离心5min,取上清溶液进行蛋白sds-page电泳分析。
1.2.6sds-page电泳分析
分别吸取10μl以上样品蛋白溶解液进行胶浓度为15%的sds-page电泳分离,sds-page凝胶配置(如下表4),具体操作方法及配置方法参照《精编分子生物学实验指南(第五版)。电泳分离结束后,取出胶体置于0.1%(w/v)考马斯亮蓝g-250染色溶液中染色45min,脱色溶液中进行洗脱,待背景脱色干净后拍照,进行蛋白含量的初步测定。
表4sds-page凝胶配制表
1.2.7蛋白诱导表达体系优化
1.2.7.1iptg最佳诱导温度的优化
将保存的阳性克隆菌群复苏,以1:100的比例接种于lb培养基(含amp),200r/min,37℃摇床中培养至d600nm=1时,加入iptg使其终浓度为1.0mmol/l,分别置于不同的温度梯度16℃、25℃、31℃、37℃、43℃条件下进行诱导表达,取10μl蛋白样品进行sds-page电泳分离,确定最佳诱导温度。
1.2.7.2iptg最佳诱导时间的优化
在最佳诱导温度条件下,确保iptg终浓度为1.0mmol/l培养条件不变下,分别在不同的时间点取样:0h、4h、8h、12h、16h,取10μl以上蛋白样品进行sds-page电泳分析,确定最佳的诱导时间。
1.2.7.3iptg最佳诱导浓度的优化
在优化得到的最佳诱导温度和培养时间基础上,分别加入不同终浓度的iptg诱导剂:0mmol/l、0.2mmol/l、0.4mmol/l、0.6mmol/l、0.8mmol/l、1.0mmol/l、1.2mmol/l、1.5mmol/l进行诱导,取10μl以上蛋白样品进行sds-page电泳分析,最终确定最佳的诱导浓度。
1.2.8蛋白的纯化及蛋白浓度测定
1.2.8.1蛋白的ni-nta亲和层析纯化
200r/min,37℃摇床中大量培养菌群至浓度为d600nm=1,加入iptg诱导剂,8000r/min离心5min,收集菌体沉淀,pbs缓冲液洗涤3次,蛋白平衡缓冲液以1/10体积重新悬起,冰浴环境下超声波完全破碎菌体,12000r/min离心5min,吸取上清作为待纯化样品。依据proteinisotmni-ntaresin操作方法进行蛋白分离纯化,将不同浓度咪唑洗脱的收集液进行sds-page电泳分析,确定咪唑的最佳纯化浓度。proteinisotmni-ntaresin对his标签蛋白进行分离纯化的过程通常包括:装柱、平衡、上样、洗涤、洗脱、再生等步骤。
1.2.8.2纯化后蛋白的浓度测定
运用bca法蛋白定量试剂盒,用酶标仪检测并绘制标准曲线,计算纯化的pet-32a(+)-plnj蛋白浓度。
1.2.9重组蛋白的westernblot鉴定
将纯化后的重组蛋白pet-32a(+)-plnj经sds-page电泳分离后,利用转印槽进行免疫印迹,随后利用一抗、二抗进行免疫杂交,最后显色发光并拍照。
2结果与分析
2.1plnj基因的pcr扩增结果
以乳酸菌基因组dna为模板,plnj基因的上游引物f和下游引物r为特异性扩增引物,以优化过的反应条件进行pcr扩增,扩增产物经核酸电泳鉴定,可见一条大小为168bp大小的特异性条带(如图1所示),与预期的片段大小相符,由此证明设计的引物可对乳酸菌的plnj基因进行特异性扩增。
2.2重组表达质粒的鉴定结果
挑选以白色单克隆菌为模板,分别以plnj基因的上游引物f和下游引物r进行pcr鉴定,扩增出184bp的特异性条带,以载体通用引物t7的上游引物f和下游引物r,可以扩增出867bp大小的目的条带(如图2所示)。
酶切鉴定结果如下图3所示,图中重组质粒单酶切的产物可见一条大小约为6000bp左右的线性质粒条带,双酶切可见多出一条约为184bp的条带,与预期的大小相符,证明了乳酸菌素基因plnj已经成功克隆至表达载体pet-32a(+)上,重组质粒的测序结果进一步证明质粒的成功构建无移码的错误。
重组蛋白的诱导表达结果
2.5.1iptg最佳诱导温度的优化
在iptg终浓度为1.0mmol/l的条件下,对pet-32a(+)-plnj重组质粒菌进行诱导12h,分析了16℃、25℃、31℃、37℃、43℃温度下诱导的这个蛋白的表达量,结果如下图4所示,蛋白在16~37℃的温度梯度中表达量都逐渐变大,在37℃的条件下表达量都达到最大,37℃后蛋白表达量都逐渐减少,因此可知,pet-32a(+)-plnj蛋白的最佳诱导温度为37℃。
2.5.2iptg最佳诱导时间的优化
在37℃,iptg终浓度为1.0mmol/l的条件下,分析了pet-32a(+)-plnj重组质粒菌分别在0h、4h、8h、12h、16h等不同的诱导时间下的蛋白表达量变化。结果如下图5所示,蛋白在诱导4h时,都开始表达目的蛋白,且目的蛋白的表达量随时间的推移逐渐提高,直到诱导12h后达到最大值,此后,蛋白的表达量渐趋于平衡。由此可知,在该条件下,iptg的最佳诱导时间为12h。
2.5.3iptg最佳诱导浓度的优化
pet-32a(+)-plnj重组质粒菌在iptg终浓度为0mmol/l时蛋白的表达很少在,而浓度在0.2~1.2mmol/l的梯度间时,蛋白的表达量有逐渐递增的趋势,在1.2mmol/l的浓度时达到最大,1.5mmol/l时不再递增,综合结果可确定pet-32a(+)-plnj重组质粒菌在37℃环境下培养12h,其最佳的iptg诱导浓度为1.2mmol/l(如图6所示)。
2.6蛋白的ni-nta亲和层析纯化结果
声破碎后的菌液通过ni-nta亲和层析柱,分别都用不同浓度(40mmol/l,80mmol/l,120mmol/l,160mmol/l,200mmol/l,240mmol/l,280mmol/l,320mmol/l)的咪唑稀释液进行洗涤纯化,并进行sds-page分析,结果显示(如图7),经过洗脱后得到的蛋白条带越来越单一,说明非特异性的蛋白杂质已明显减少,但在不同的咪唑浓度,得到的蛋白浓度及纯度不一样,以280mm的咪唑浓度的洗脱液纯化效率最高。
2.7western-blot鉴定结果
结果显示(如图8所示),在针对his•tag标签蛋白的westernblot分析中得知,pet-32a(+)-plnj重组质粒菌组和pet-32a(+)空载体菌组分别在大小约为26.0ku和20ku出现一条特异性的条带。
2.8pet-32a(+)-plnj重组乳酸菌素对四种指示菌的抑菌结果
pet-32a(+)-plnj重组乳酸菌素对四种指示菌的抑菌效果(如表5所示),pet-32a(+)-plnj重组乳酸菌素浓度为0.10mg/ml时对金黄色葡萄球菌、大肠杆菌、沙门氏菌和巴氏杆菌具有抑菌效果,可见,pet-32a(+)-plnj重组乳酸菌素具有广谱的抑菌活性。
表5pet-32a(+)-plnj重组乳酸菌素对指示菌抑菌圈的大小(mm)
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
sequencelisting
<110>福建农林大学
<120>一种提高乳酸菌素plnj的产量的基因工程方法
<130>4
<160>4
<170>patentinversion3.3
<210>1
<211>28
<212>dna
<213>人工序列
<400>1
gcggatccatgactgtgaacaaaatgat28
<210>2
<211>28
<212>dna
<213>人工序列
<400>2
atgtcgacttaacgacggattgctctgc28
<210>3
<211>20
<212>dna
<213>人工序列
<400>3
taatacgactcactataggg20
<210>4
<211>19
<212>dna
<213>人工序列
<400>4
gctagttattgctcagcgg19
- 还没有人留言评论。精彩留言会获得点赞!