载银纳米复合抗菌材料及制备方法与流程
本发明属于有机多孔材料领域,特别涉及一种载银纳米复合抗菌材料的制备方法。
背景技术:
近年来,共轭微孔聚合物(conjugatedmicroporouspolymers,cmps)材料的合成及应用研究一直备受关注。作为一类重要的有机多孔材料,共轭微孔聚合物以其自身的共轭刚性结构和永久的微孔性质,不仅在吸附,分离,多相催化等传统领域具有应用价值,同时,在光电,储能,传感等领域也有潜在应用。设计思路从最初的选取不同单体,不同合成方法构筑cmps转变为cmps的后修饰和官能团化,逐步有了更多新的应用领域。
作为一类重要的氮杂稠环,1,10-菲罗啉及其衍生物在在络合金属离子、光电器件等方面具有重要价值。
无机抗菌剂,即多孔材料负载本身具抗菌效果的银,铜等金属的抗菌剂,以其耐热性好,不产生抗药性,毒性低等优于天然抗菌剂和有机抗菌剂的特点而受到广泛研究。
目前,亟需一种新型的cmps并将其应用于抗菌载体。
技术实现要素:
本发明的目的是克服现有技术的不足,提供共轭微孔聚合物sn1-cmp或sn2-cmp。
本发明的第二个目的是提供共轭微孔聚合物sn1-cmp或sn2-cmp的制备方法。
本发明的第三个目的是提供用共轭微孔聚合物sn1-cmp或sn2-cmp为原料制备的载银纳米复合抗菌材料。
本发明的第四个目的是提供载银纳米复合抗菌材料的制备方法。
本发明的技术方案概述如下:
共轭微孔聚合物sn1-cmp或sn2-cmp,sn1-cmp用式i所示,sn2-cmp用式iv所示;
式i是由化合物iii-br和化合物ii,通过a2b4型sonogashira偶联反应制成:
式iv是由化合物v-br和化合物ii,通过a2b4型sonogashira偶联反应制成:
其中r1为叔丁基。
共轭微孔聚合物sn1-cmp或sn2-cmp的制备方法,包括如下步骤:
在氩气氛围下,将钯催化剂、配体加入到有机溶剂中,室温条件下搅拌均匀,加入化合物iii-br或v-br、化合物ii、碘化亚铜、三乙胺,冷冻除氧2-4次,加热至80-120℃,反应24~72小时,冷却至室温,过滤,洗涤,干燥,得到共轭微孔聚合物sn1-cmp或sn2-cmp;
所述sn1-cmp的制备见反应式1:
反应式1:
所述sn2-cmp的制备见反应式2:
反应式2:
其中r1为叔丁基。
钯催化剂优选二氰基苯二氯化钯或四三苯基膦钯,还可以选用其它的钯催化剂进行催化。
配体优选三(邻甲苯基)膦或三苯基膦。
有机溶剂为n,n-二甲基甲酰胺或四氢呋喃,也可以选用其它的有机溶剂。
载银纳米复合抗菌材料的制备方法,包括如下步骤:
将银盐溶于醇中,加入共轭微孔聚合物sn1-cmp或sn2-cmp,室温下,避光搅拌8-12小时,过滤,用醇洗涤,得到固体,放入装有甲醇的索氏提取器中,洗涤去除未吸附的银盐,干燥,得到载银纳米复合抗菌材料ag-sn1-cmp或ag-sn2-cmp。
银盐与共轭微孔聚合物sn1-cmp或sn2-cmp的质量比优选2:1。
上述银盐优选四氟硼酸银或硝酸银。
醇为甲醇或乙醇,也可以采用其它的醇。
上述方法制备的载银纳米复合抗菌材料。
本发明的优点:
本发明合成了两种共轭微孔聚合物sn1-cmp或sn2-cmp,并以这两种共轭微孔聚合物为载体,负载银离子,制备得到载银纳米复合抗菌材料ag-sn1-cmp或ag-sn2-cmp。扩展了共轭微孔聚合物材料的应用范围,载银纳米复合抗菌材料ag-sn1-cmp或ag-sn2-cmp抗菌效果好,在生物抗菌领域具有潜在应用价值。
附图说明
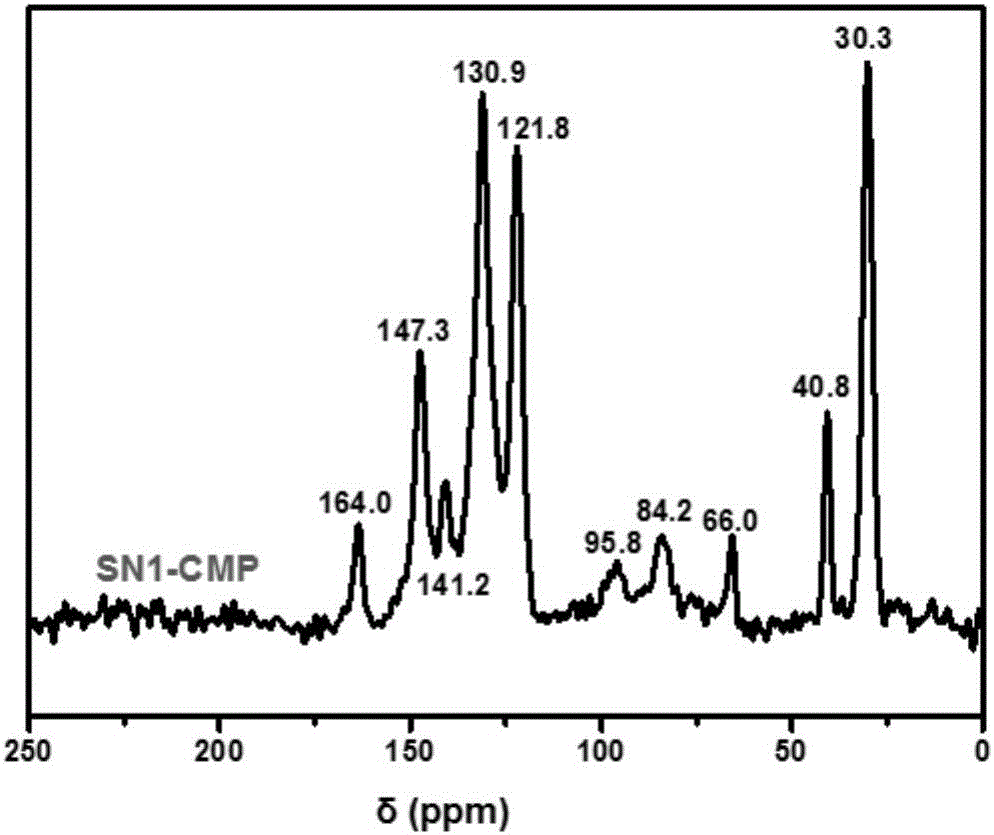
图1为共轭微孔聚合物sn1-cmp的固体核磁图。
图2为共轭微孔聚合物sn2-cmp的固体核磁图。
图3为共轭微孔聚合物sn1-cmp与其合成单体的红外光谱图。
图4为共轭微孔聚合物sn2-cmp与其合成单体的红外光谱图。
图5为共轭微孔聚合物sn1-cmp的扫描电镜图(sem)。
图6为共轭微孔聚合物sn2-cmp的扫描电镜图(sem)。
图7为共轭微孔聚合物sn1-cmp的透射电镜图(tem)。
图8为共轭微孔聚合物sn2-cmp的透射电镜图(tem)。
图9为载银纳米复合抗菌材料ag-sn1-cmp的透射电镜图(tem)。
图10为载银纳米复合抗菌材料ag-sn2-cmp的透射电镜图(tem)。
图11为共轭微孔聚合物sn1-cmp的n2吸附与脱附曲线。
图12为共轭微孔聚合物sn2-cmp的n2吸附与脱附曲线。
图13为载银纳米复合抗菌材料ag-sn1-cmp的抗菌实验。
图14为载银纳米复合抗菌材料ag-sn2-cmp的抗菌实验。
具体实施方式
下面结合具体实施例对本发明作进一步的说明。为了便于理解本发明,以下的描述是对本发明的具体说明,不应看作对本发明的限定。
实施例1
单体sn1-br(iii-br)的合成:(实施例1的制备仅是举例,对本发明不作任何限定)
(1)将化合物a1(16.79mmol,4.5g)、叔丁酰氯(31.45mmol,4.23g)、三乙胺(4ml),加入到100ml四氢呋喃溶液中,室温搅拌12小时。减压旋出溶剂,加入二氯甲烷和水进行萃取洗涤,再用无水硫酸镁进行干燥,最后旋干。进行柱层析提纯,洗脱剂为二氯甲烷:石油醚=2:3,得白色固体化合物b1(6.17g,84%)。
(2)将化合物b1(2.29mmol,1g)、苯硼酸酯(5.28mmol,1.11g)、碳酸氢钠(95.22mmol,8g)、四(三苯基膦)钯(0.07mmol,0.08g)加入到250ml的二口瓶中,抽气脱气三次,加入四氢呋喃(75ml)和水(30ml)的混合溶液中,抽气脱气三次,最终在氮气保护下,加热到70℃,回流24小时。冷却至室温,减压旋出溶剂,加入二氯甲烷和水进行萃取洗涤,再用无水硫酸镁进行干燥,最后旋干。进行柱层析提纯,洗脱剂为二氯甲烷:石油醚=1:1,得白色固体化合物c1(0.61g,61%)。
(3)将化合物c1(1.13mmol,0.5g)、三氯氧磷(15ml)、五氧化二磷(23.25mmol,3.3g)加入到50ml单口圆底烧瓶中,加热到116℃,回流30小时。冷却至室温后,缓慢倒入冰水中,并用氢氧化钾调节ph至10,边调节边冷却。加入二氯甲烷进行萃取,再用无水硫酸镁干燥,最后旋干。过中性氧化铝柱,最终,用二氯和石油醚重结晶,得淡黄色固体化合物sn1(0.32g,71%)。
(4)将二异丙胺(1ml)加入到四氢呋喃(20ml)中,冷冻除氧后,在-78℃条件下,缓慢加入正丁基锂(2.5ml),维持低温1小时,取出,室温搅拌1小时,溶液呈黄色;然后,-78℃下,加sn1(0.25mmol,0.1g),维持低温1小时,取出,室温搅拌1小时,溶液呈绿色;然后,-78℃下,加入四溴化碳(2.6mmol,0.86g),自然回温,过夜,溶液呈棕色。加水淬灭反应,旋出溶剂,加入二氯甲烷进行萃取,再用无水硫酸镁干燥,最后旋干。过中性氧化铝柱,最终,用二氯和石油醚重结晶,得纯白色固体化合物sn1-br(iii-br)(0.12g,84%),见下式:
其中,r1为叔丁基。
实施例2
单体sn2-br(v-br)的合成:(实施例2的制备仅是举例,对本发明不作任何限定)
(1)将化合物a2(3.6mmol,1g)、叔丁酰氯(8.6mmol,1.05g)、三乙胺(3ml),加入到35ml二氯甲烷溶液中,室温搅拌24小时。冷却至室温,倒入水中,加入乙酸乙酯和水进行萃取洗涤,再用无水硫酸镁进行干燥,最后旋干。进行柱层析提纯,洗脱剂为二氯甲烷,得白色固体化合物b2(1.5g,93%);
(2)将化合物b2(2.24mmol,1g)加入到30mldmf中,随后,逐滴加入溶有n-溴代丁二酰亚胺(5mmol,0.9g)的30mldmf溶液,加完后,加热至50℃,过夜。冷却至室温,倒入水中,用二氯甲烷萃取,无水硫酸镁干燥,然后旋干。进行柱层析提纯,洗脱剂为二氯甲烷,得淡黄色固体化合物c2(1.04g,77%)。
(3)将c2(0.16mmol,0.1g)、三氯氧磷(8ml)、五氧化二磷(4.95mmol,0.71g)加入到50ml单口圆底烧瓶中,加热到116℃,回流30小时。冷却至室温后,缓慢倒入冰水中,并用氢氧化钾调节ph至10,边调节边冷却。加入二氯甲烷进行萃取,再用无水硫酸镁干燥,最后旋干。过中性氧化铝柱,最终,用二氯和石油醚重结晶,得灰白色固体化合物sn2-br(v-br)(0.085g,91%)。
其中,r1为叔丁基。
实施例3
共轭微孔聚合物sn1-cmp(i)的制备
在氩气氛围下,将pdcl2(phcn)2(0.0075mmol,2.88mg)和三(邻甲苯基)膦(0.015mmol,4.56mg)加入到4mldmf中,室温搅拌10min后,加入sn1-br(iii-br)(0.1mmol,56.44mg),四(4-乙炔基苯基)-甲烷(tpm,化合物ii),(0.075mmol,31.45mg),碘化亚铜(0.00075mmol,0.14mg),三乙胺(2ml),冷冻除氧3次,加热至100℃,反应72小时。冷却至室温,过滤,依次用三氯甲烷,丙酮,甲醇,水洗涤,重复三次,除去未反应的单体和催化剂,过滤出的固体放入真空干燥箱中,设置80℃,干燥24小时,得黄色粉末(67mg,产率87%)。
图1为本实施例的共轭微孔聚合物sn1-cmp(i)的固体核磁碳谱图,其中,164.0-121.8ppm为芳香碳,95.8和84.2ppm为碳碳三键的碳,证明了两个单体的有效连接,66.0ppm为四(4-乙炔基苯基)-甲烷(tpm)的季碳,40.8ppm为叔丁基的季碳,30.3ppm为甲基碳。该测试是在brukerwbavanceii400mhz的核磁光谱仪上进行的。
单体sn1-br与四(4-乙炔基苯基)-甲烷(tpm)及本实施例得到的共轭微孔聚合物(sn1-cmp)的红外光谱图如图3所示,由图可知,其中,tpm在3283cm-1处的吸收峰为炔基氢的伸缩振动峰,而在得到的共轭微孔聚合物的红外曲线中该处峰消失,说明反应之后炔基氢消失,同时,单体sn1-br在495cm-1处的c-br吸收峰在共轭微孔聚合物红外曲线中消失,以上两点均验证了本实施例产物共轭微孔聚合物的结构。该红外光谱测试采用brukeralphaspectrometer红外光谱仪,压片制样,共轭微孔聚合物事先进行干燥处理。
共轭微孔聚合物sn1-cmp(i)的扫描电镜图(sem)为图5和透射电镜图(tem)为图7,可看出质地均匀的多孔结构。图11的比表面积分析,sn1-cmp的比表面积为1003m2·g-1,nldft拟合平均孔径大小约1.4nm,以微孔为主要孔道大小。扫描电镜使用的是hitachilimitedmodels-4800microscope,透射电镜使用的是jeolmodeljem-2100fmicroscope以及比表面积分析采用beljapaninc.modelbelsopr-miniiianalyser。
用四三苯基膦钯替代本实施例的pdcl2(phcn)2、用三苯基膦替代本实施例的三(邻甲苯基)膦,用四氢呋喃替代本实施例的dmf,用冷冻除氧2次替代冷冻除氧3次,加热至80℃替代100℃,用反应24小时,替代反应72,其它同本实施例,制备出共轭微孔聚合物sn1-cmp。
实施例4
共轭微孔聚合物sn2-cmp(iv)的制备
在氩气氛围下,将pdcl2(phcn)2(0.0075mmol,2.88mg)和三(邻甲苯基)膦(0.015mmol,4.56mg)加入到4mldmf溶液中,室温搅拌10min,加入sn2-br(v-br)(0.1mmol,56.84mmol),四(4-乙炔基苯基)-甲烷(tpm,化合物ii)(0.075mmol,31.45mg),碘化亚铜(0.00075mmol,0.14mg),三乙胺(2ml),冷冻除氧3次,加热至100℃,反应72小时。冷却至室温,过滤,依次用三氯甲烷,丙酮,甲醇,水洗涤,重复三次,除去未反应的单体和催化剂,过滤出的固体放入真空干燥箱中,设置80℃,干燥24小时,得黄色粉末(73mg,产率94%)。
图2为本实施例的共轭微孔聚合物sn2-cmp(iv)的固体核磁碳谱图,其中,163.6-121.8ppm为芳香碳,98.1和83.8ppm为碳碳三键的碳,证明了两个单体的有效连接,65.8ppm为四(4-乙炔基苯基)-甲烷(tpm)的季碳,39.9ppm为叔丁基的季碳,30.0ppm为甲基碳。该测试是在brukerwbavanceii400mhz的核磁光谱仪上进行的。
单体sn2-br与四(4-乙炔基苯基)-甲烷(tpm)及本实施例得到的共轭微孔聚合物(sn2-cmp)的红外光谱图如图4所示,由图可知,其中,tpm在3283cm-1处的吸收峰为炔基氢的伸缩振动峰,而在得到的共轭微孔聚合物的红外曲线中该处峰消失,说明反应之后炔基氢消失,同时,单体sn2-br在491cm-1处的c-br吸收峰在共轭微孔聚合物红外曲线中消失,以上两点均验证了本实施例产物共轭微孔聚合物的结构。该红外光谱测试采用brukeralphaspectrometer红外光谱仪,压片制样,共轭微孔聚合物事先进行干燥处理。
轭微孔聚合物sn2-cmp(iv)的扫描电镜图(sem)为图6和透射电镜图(tem)为图8,可看出质地均匀的多孔结构。图12的比表面积分析,sn2-cmp的比表面积为925m2·g-1,nldft拟合平均孔径大小为1.4nm,以微孔为主要孔道大小。扫描电镜使用的是hitachilimitedmodels-4800microscope,透射电镜使用的是jeolmodeljem-2100fmicroscope以及比表面积分析采用beljapaninc.modelbelsopr-miniiianalyser。
用四三苯基膦钯替代本实施例的pdcl2(phcn)2、用三苯基膦替代本实施例的三(邻甲苯基)膦,用四氢呋喃替代本实施例的dmf,用冷冻除氧4次替代冷冻除氧3次,加热至120℃替代100℃,用反应48小时,替代反应72小时,其它同本实施例,制备出共轭微孔聚合物sn2-cmp。
实施例5
载银纳米复合抗菌材料ag-sn1-cmp的制备方法,包括如下步骤:
将四氟硼酸银(10mg)溶解于5ml无水甲醇,加入共轭微孔聚合物sn1-cmp(5mg),室温下,避光搅拌8小时,用甲醇反复洗涤,除去未吸附的四氟硼酸银,分离,得到固体,放入装有甲醇的索氏提取器中,洗涤去除未吸附的银盐,干燥,得到载银纳米复合抗菌材料ag-sn1-cmp。
银的质量分数通过icp-ms确定,为12.32wt%(ag-sn1-cmp)。
图9为ag-sn1-cmp的透射电镜图,可以看到银纳米粒子的存在。透射电镜使用的是jeolmodeljem-2100fmicroscope。
经典抗菌琼脂实验测试如图13,在直径为80mm的琼脂培养皿中培养等量的大肠杆菌菌落,放置相同时间条件下,随着加入抗菌剂浓度的增加,细菌死亡率增加,即抗菌能力增强。
实验证明,用硝酸银替代本实施例的四氟硼酸银,用乙醇替代本实施例的甲醇,其它同本实施例,可以制备出载银纳米复合抗菌材料ag-sn1-cmp,并有相似的抗菌效果。
实施例6
载银纳米复合抗菌材料ag-sn2-cmp的制备方法,包括如下步骤:
将四氟硼酸银(10mg)溶解于5ml无水甲醇,加入共轭微孔聚合物sn2-cmp(5mg),室温下,避光搅拌12小时,用甲醇反复洗涤,除去未吸附的四氟硼酸银,得到固体,放入装有甲醇的索氏提取器中,洗涤去除未吸附的银盐,干燥,得到载银纳米复合抗菌材料ag-sn2-cmp。
银的质量分数通过icp-ms确定,为6.22wt%(ag-sn2-cmp)。
图10为ag-sn2-cmp的投射电镜图,可以看到银纳米粒子的存在。透射电镜使用的是jeolmodeljem-2100fmicroscope。
经典抗菌琼脂实验测试如图14,在直径为80mm的琼脂培养皿中培养等量的大肠杆菌菌落,放置相同时间条件下,随着加入抗菌剂浓度的增加,细菌死亡率增加,即抗菌能力增强。
实验证明,用硝酸银替代本实施例的四氟硼酸银,用乙醇替代本实施例的甲醇,其它同本实施例,可以制备出载银纳米复合抗菌材料ag-sn2-cmp,并有相似的抗菌效果。
通过琼脂抗菌实验,将不同浓度的ag-sn-cmp分散于琼脂细菌培养皿中,静置观察抗菌情况。放置相同时间条件下,随着加入抗菌剂浓度的增加,抗菌效果增强。
- 还没有人留言评论。精彩留言会获得点赞!