一种1/2水头孢唑肟钠化合物的制作方法
本发明属于化学工程医药结晶
技术领域:
,具体涉及一种1/2水头孢唑肟钠化合物及其制法。
背景技术:
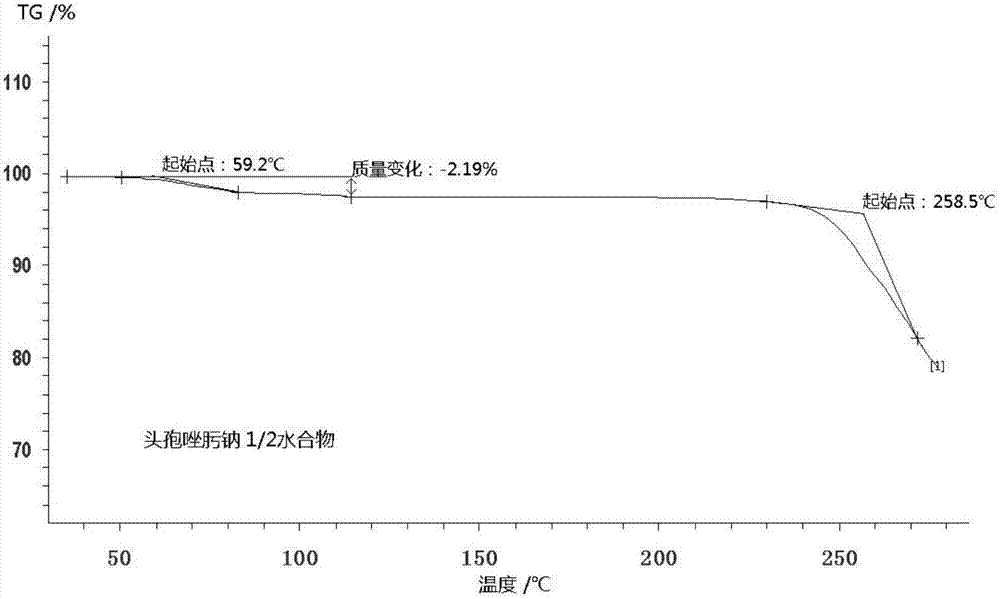
:头孢唑肟钠最早由日本藤泽药品工业株式会社研制开发,并于1982年首先在日本上市,商品名epocelin,为属第3代头孢菌素抗生素,其作用机制是通过抑制细菌细胞壁粘肽的生物合成而达到杀菌作用,具有广谱、高效、耐酶、低毒和能透过血脑屏障的特点。头孢唑肟钠对各种革兰氏阳性菌和革兰氏阴性菌产生的光谱β-内酰胺酶稳定;对大肠埃希菌、肺炎克雷伯菌、奇异变形杆菌等肠杆菌科细菌有强大抗菌作用,绿铜假单胞菌等假单胞菌属和不动杆菌属对本品敏感性差;对流感嗜血杆菌和淋病奈瑟球菌有良好抗菌作用;对金黄色葡萄球菌和表皮葡萄球菌的作用较第一、第二代头孢菌素为差,耐甲氧西林金黄色葡萄球菌和肠球菌属对本品耐药,各种链球菌对本品均高度敏感;消化球菌、消化链球菌和部分拟杆菌属等厌氧菌对本品多呈敏感。临床上头孢唑肟钠用于治疗敏感菌所致的下呼吸道感染、尿路感染、腹腔感染、盆腔感染、败血症、皮肤软组织感染、骨和关节感染、肺炎链球菌或流感嗜血杆菌所致脑膜炎和单纯性淋病。有关头孢唑肟钠的合成,文献报道了三条合成路线,(一)以7-苯乙酰氨基-3-头孢菌素-4-羧酸对甲氧苄酯为原料,经多次脱去保护基团,再与成盐剂制得头孢唑肟钠,此路线反应步骤长,增加了操作程序及生产成本,且副产物较多。(二)以7-氨基-3-去甲基-3-头孢烷酸(7-anca)为原料与氨噻肟乙酸直接缩合,然后再成盐,此路线副反应较多,所得产物需经氧化铝层析柱纯化,还要使用大量的洗脱剂,成本高、操作复杂且对环境造成污染,不适合工业化生产。(三)以7-anca为原料,与2-(2-氨基-4-噻唑基)-2-甲氧亚胺-乙酰-苯并噻唑硫酯缩合制得头孢唑肟酸,再与成盐剂反应得头孢唑肟钠。以上方法制备的头孢唑肟钠化合物,存在着储存稳定性差、热分解温度低、流动性差、易吸湿等等严重问题。从而影响产品质量,造成制剂产品不澄清,浊度不合格,并且降低了制剂的稳定性,为了获得一种性能更为优良的头孢唑肟钠化合物,特提出本发明。技术实现要素:本发明公开了一种头孢唑肟钠新的溶剂化物,更具体的为1/2水头孢唑肟钠化合物,即每摩尔头孢唑肟钠化合物含1/2摩尔水,分子式c13h12n5nao5s2·1/2h2o,分子量414.38,结构式为:卡尔费休法是各种测定物质中含有水分方法中最为专一、准确的方法之一,已被列为许多物质中水分测定的基本方法,尤其是对有机化合物,结果准确可靠。本发明公开的1/2水头孢唑肟钠化合物用卡尔费休法测定水分含量在2.13%-2.22%之间,1/2水头孢唑肟钠化合物理论水分含量为2.17%,可确定本发明头孢唑肟钠化合物含有1/2摩尔水。本发明所述的1/2水头孢唑肟钠化合物,其tg分析结果显示,失重百分率经计算可知约为2.19%,头孢唑肟钠分子中水的理论百分含量是2.17%,参照费休氏法测得头孢唑肟钠水分含量为2.13~2.22%,而实验测得tg失重为2.19%,与理论水含量基本相符。因此可推断头孢唑肟钠tg失重是脱除水所致,且每摩尔头孢唑肟钠含1/2摩尔水。如附图1所示。数据由热分析-质谱联用仪(netzschsta449c)分析得到。分析条件为:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min,升温速率10k/min,温度测试范围为25~400℃。样品开始分解温度为59.2℃,温度高于258.5℃,样品快速分解。本发明所述的1/2水头孢唑肟钠化合物,其粉末x射线衍射图谱在衍射角2θ为11.43±0.2°,13.24±0.2°,16.54±0.2°,17.85±0.2°,19.46±0.2°,20.66±0.2°,22.33±0.2°,23.30±0.2°,23.65±0.2°,24.60±0.2°处有特征衍射峰,衍射角的相对衍射强度分别为89.67,39.41,19.06,46.51,100,22.11,46.76,38.69,40.87,24.76。如附图2所示。x射线粉末衍射测试条件:荷兰panalytical公司的empyrean(锐影)x射线衍射仪,cukα辐射,光管电压40kv,灯丝电流300ma,连续扫描,步长0.02°,扫描速度8°/min,扫描范围为2~50°。文献多有报道相同物质的不同晶型、相同物质的不同溶剂化物有相同的粉末x射线衍射谱图或者有部分相同的粉末x射线衍射谱图,所以有必要根据《多晶型药品的质量控制技术与方法指导原则》,给出其他的鉴别方法证明本专利报道的是新的水合物。本发明所述的1/2水头孢唑肟钠化合物,其傅里叶变换红外光谱在波数为3425.9±2cm-1,3239.6±2cm-1,1746.3±2cm-1,1648.1±2cm-1,1537.9±2cm-1,1414.7±2cm-1,1365.2±2cm-1,1180.9±2cm-1,1034.0±2cm-1,992.5±2cm-1,811.0±2cm-1处有特征吸收峰,如附图3所示。红外光谱测试条件为:安捷伦cary630,溴化钾压片。本发明所述的1/2水头孢唑肟钠化合物,其dsc分析结果显示,在约73.9℃有吸热峰,在约264.9℃有放热峰。如附图4所示。dsc数据由热分析-质谱联用仪(netzschsta449c)分析得到,分析条件为:样品2~10mg,氧化铝坩埚,高纯氮气做反应气和保护气,流量分别为40ml/min和30ml/min。升温速率10℃/min,温度范围25~400℃。本发明所述的1/2水头孢唑肟钠化合物,具体制备方法包括:(1)将7-anca加入三氯甲烷中,搅拌成混悬液,缓慢加入三乙胺,搅拌,再加入ae-活性酯反应,反应完毕,降低温度,加入水提取,分离水相,加入活性炭,搅拌吸附,过滤,洗涤,合并滤液,用盐酸溶液调节滤液ph值,搅拌结晶,过滤,三氯甲烷洗涤,真空干燥,得头孢唑肟酸;(2)将头孢唑肟酸加入纯化水中,搅拌成混悬液,降低温度,缓慢加入三乙胺,搅拌溶解,加活性炭搅拌脱色,过滤,纯化水洗涤,保持温度,缓慢加入异辛酸钠的乙醇水溶液,搅拌,加乙醇结晶,缓慢搅拌结晶,过滤,乙醇洗涤,真空干燥,得1/2水头孢唑肟钠化合物。上述制备方法中,步骤(1)中所述的加入ae-活性酯后的反应温度为30-35℃,所述的加入ae-活性酯后反应时间为4-5h,所述的降低降温至10-15℃,所述的加入盐酸溶液调节ph值至2.0-2.5,所述的结晶温度为10-15℃,所述的结晶时间为1-2h,所述的干燥温度为30-40℃。上述制备方法中,步骤(2)中所述的降低温度至5-10℃,所述的乙醇水溶液为50%-70%的乙醇水溶液,所述的头孢唑肟酸与异辛酸钠的反应时间为30min-1h,所述的搅拌结晶时间为1-2h,所述的干燥温度为30-40℃。本发明提供的1/2水头孢唑肟钠化合物的制备方法优点为收率高、成本低、产品品质稳定;所制备的1/2水头孢唑肟钠化合物高效液相色谱含量符合药典规定。本发明提供的1/2水头孢唑肟钠化合物热稳定性好,差示热分析表明其在约73.9℃有吸热峰,在约264.9℃有放热峰。加速稳定性试验表明本发明1/2水头孢唑肟钠化合物的稳定性优于头孢唑肟钠无水物。本发明提供的1/2水头孢唑肟钠化合物与头孢唑肟钠无水物相比更不易吸湿。因此本发明提供的1/2水头孢唑肟钠化合物在稳定性、抗湿性方面优于头孢唑肟钠无水物,具有更广泛的应用前景。本发明的进一步的目的,提供了一种含1/2水头孢唑肟钠化合物的药物组合物。优选地,所述药物组合物包含1/2水头孢唑肟钠化合物和药学上接受的赋形剂。更优选地,药物组合物选自药学上可接受的剂型。本发明的进一步的目的,提供了一种1/2水头孢唑肟钠化合物及其药物组合物在制备抗菌药物中的应用。附图说明图11/2水头孢唑肟钠化合物的tg图;图21/2水头孢唑肟钠化合物的x射线粉末衍射谱(xrd)图;图31/2水头孢唑肟钠化合物的傅里叶变换红外光谱(ft-ir)图;图41/2水头孢唑肟钠化合物的差热分析(dsc)图。具体实施方式下面将通过具体实施方式对本发明做进一步说明,但并不因此将本发明限制在所述的实施例范围中,本领域的技术人员应理解,对本
发明内容所做的等同替换,或相应的改进,仍属于本发明的保护范围之内。实施例11/2水头孢唑肟钠化合物的制备(1)将100g7-anca加入2l三氯甲烷中,搅拌成混悬液,缓慢加入200ml三乙胺,搅拌20min,再加入193gae-活性酯30℃反应4h,反应完毕,降低温度至10℃,加入4l水提取,分离水相,加入20g活性炭,搅拌吸附30min,过滤,洗涤,合并滤液,用2mol/l的盐酸溶液调节ph值至2.0,10℃搅拌结晶1h,过滤,三氯甲烷洗涤,30℃真空干燥20分钟,得头孢唑肟酸169.3g;(2)将头孢唑肟酸加入500ml纯化水中,搅拌成混悬液,降低温度至5℃,缓慢加入70ml三乙胺,搅拌溶解,加入5g活性炭搅拌脱色20min,过滤,纯化水洗涤,缓慢加入溶有100g异辛酸钠的50%的乙醇水溶液200ml,搅拌反应30min,加入5l乙醇,缓慢搅拌结晶1h,过滤,乙醇洗涤,30℃真空干燥30分钟,得1/2水头孢唑肟钠化合物161.1g。x射线粉末衍射图谱在衍射角2θ为11.43°,13.24°,16.54°,17.85°,19.46°,20.66°,22.33°,23.30°,23.65°,24.60°处有特征衍射峰,衍射角的相对衍射强度分别为89.67,39.41,19.06,46.51,100,22.11,46.76,38.69,40.87,24.76。傅里叶红外光谱在波数为3425.9cm-1,3239.6cm-1,1746.3cm-1,1648.1cm-1,1537.9cm-1,1414.7cm-1,1365.2cm-1,1180.9cm-1,1034.0cm-1,992.5cm-1,811.0cm-1处有特征峰,各个波数对应的特征峰基本一致,都在范围之内。hplc法检测纯度为99.69%;卡尔费休法测定水分为2.17%,热重分析失重为2.19%,这样与含有的1/2个水的结果(理论值2.17%)基本一致;元素分析理论值为:c:37.68%,h:3.16%,n:16.90%,na:5.55%,o:21.24%,s:15.47%;实测值为:c:37.63%,h:3.17%,n:16.92%,na:5.54%,o:21.25%,s:15.49%。实施例21/2水头孢唑肟钠化合物的制备(1)将120g7-anca加入2.5l三氯甲烷中,搅拌成混悬液,缓慢加入240ml三乙胺,搅拌20min,再加入232gae-活性酯35℃反应5h,反应完毕,降低温度至15℃,加入5l水提取,分离水相,加入20g活性炭,搅拌吸附30min,过滤,洗涤,合并滤液,用2mol/l的盐酸溶液调节ph值至2.5,10℃搅拌结晶1h,过滤,三氯甲烷洗涤,30℃真空干燥25分钟,得头孢唑肟酸211.8g;(2)将头孢唑肟酸加入500ml纯化水中,搅拌成混悬液,降低温度至10℃,缓慢加入100ml三乙胺,搅拌溶解,加入5g活性炭搅拌脱色20min,过滤,纯化水洗涤,缓慢加入溶有125g异辛酸钠的70%的乙醇水溶液250ml,搅拌反应1h,加入6l乙醇,缓慢搅拌结晶2h,过滤,乙醇洗涤,40℃真空干燥20分钟,得1/2水头孢唑肟钠化合物207.3g。x射线粉末衍射图谱在衍射角2θ为11.44°,13.20°,16.57°,17.86°,19.42°,20.68°,22.35°,23.33°,23.66°,24.62°处有特征衍射峰,衍射角的相对衍射强度分别为88.40,39.89,19.77,45.50,100,23.09,45.19,37.63,39.28,25.70。傅里叶红外光谱在波数为3426.3cm-1,3239.9cm-1,1746.6cm-1,1648.2cm-1,1537.5cm-1,1414.4cm-1,1365.6cm-1,1180.3cm-1,1034.5cm-1,992.7cm-1,811.2cm-1处有特征峰,各个波数对应的特征峰基本一致,都在范围之内。hplc法检测纯度为99.72%;卡尔费休法测定水分为2.13%,热重分析失重为2.16%,这样与含有的1/2个水的结果(理论值2.17%)基本一致;元素分析理论值为:c:37.68%,h:3.16%,n:16.90%,na:5.55%,o:21.24%,s:15.47%;实测值为:c:37.66%,h:3.16%,n:16.91%,na:5.57%,o:21.23%,s:15.47%。实施例31/2水头孢唑肟钠化合物的制备(1)将90g7-anca加入1.5l三氯甲烷中,搅拌成混悬液,缓慢加入180ml三乙胺,搅拌20min,再加入174gae-活性酯35℃反应4.5h,反应完毕,降低温度至12℃,加入4l水提取,分离水相,加入20g活性炭,搅拌吸附20min,过滤,洗涤,合并滤液,用2mol/l的盐酸溶液调节ph值至2.2,12℃搅拌结晶1.5h,过滤,三氯甲烷洗涤,35℃真空干燥20分钟,得头孢唑肟酸152.4g;(2)将头孢唑肟酸加入400ml纯化水中,搅拌成混悬液,降低温度至8℃,缓慢加入90ml三乙胺,搅拌溶解,加入5g活性炭搅拌脱色20min,过滤,纯化水洗涤,缓慢加入溶有90g异辛酸钠的60%的乙醇水溶液200ml,搅拌反应1h,加入4l乙醇,缓慢搅拌结晶1.5h,过滤,乙醇洗涤,35℃真空干燥25分钟,得1/2水头孢唑肟钠化合物149.6g。x射线粉末衍射图谱在衍射角2θ为11.40°,13.22°,16.55°,17.89°,19.45°,20.66°,22.31°,23.35°,23.64°,24.68°处有特征衍射峰,衍射角的相对衍射强度分别为89.52,38.17,20.25,43.91,100,22.63,44.31,36.09,38.74,24.99。傅里叶红外光谱在波数为3426.0cm-1,3239.2cm-1,1747.1cm-1,1648.5cm-1,1538.2cm-1,1414.6cm-1,1365.2cm-1,1180.5cm-1,1034.3cm-1,992.2cm-1,811.0cm-1处有特征峰,各个波数对应的特征峰基本一致,都在范围之内。hplc法检测纯度为99.66%;卡尔费休法测定水分为2.22%,热重分析失重为2.20%,这样与含有的1/2个水的结果(理论值2.17%)基本一致;元素分析理论值为:c:37.68%,h:3.16%,n:16.90%,na:5.55%,o:21.24%,s:15.47%;实测值为:c:37.70%,h:3.15%,n:16.88%,na:5.56%,o:21.25%,s:15.46%。对比例1头孢唑肟钠化合物的制备参照文献《黑龙江科技信息》报道中《关于头孢唑肟钠的合成工艺的探讨》的方法制备头孢唑肟钠化合物(1)将120g7-anca加入3l异丙醇中,搅拌,再加入232gae-活性酯,搅拌均匀,缓慢加入2500ml三乙胺,反应液澄清后,继续反应1h,加入4l水提取,分离水相,水相用异丙醇洗涤,加入20g活性炭,搅拌吸附20min,过滤,滤液采用al2o3柱层析后水层中加入1.2l丙酮,缓慢加入2mol/l的盐酸溶液,结晶液浑浊时开始养晶30min,继续加2mol/l的盐酸溶液调节ph值至2.5,8℃搅拌结晶1h,过滤,乙酸乙酯洗涤,40℃真空干燥20分钟,得头孢唑肟酸164.1g;(2)将头孢唑肟酸加入500ml纯化水中,加入100g无水乙酸钠搅拌至溶液澄清,加入6g活性炭搅拌脱色30min,过滤,纯化水洗炭饼,滤液中加丙酮至结晶析出,缓慢搅拌养晶1h,继续加丙酮至15l,降温至10℃,搅拌结晶1h,过滤,丙酮洗涤,40℃真空干燥50分钟,得头孢唑肟钠化合物171.5g。hplc法检测纯度为99.21%;卡尔费休法测定水分为4.31%,热重分析失重为4.40%;元素分析理论值为:c:38.52%,h:2.98%,n:17.28%,na:5.67%,o:19.73%,s:15.82%;实测值为:c:38.50%,h:2.99%,n:17.30%,na:5.64%,o:19.74%,s:15.83%。对比例2头孢唑肟钠1.75水合物的制备采用专利cn101781316a中实施例1的方法制备的头孢唑肟钠1.75水合物在500ml三角瓶中加头孢唑肟酸100g、水200ml,搅拌使成悬浮液,在5℃下滴加三乙胺40ml,搅拌溶解,加活性炭1g,搅拌30分钟,抽滤,水洗,抽滤,5℃下在滤液中滴加200ml异辛酸钠和乙醇混合液(含异辛酸钠65g),搅拌,冰醋酸调节ph至7.0,缓慢滴加丙酮500ml和乙醇1500ml混合液,5℃以下放置,使固体充分析出,抽滤,少量乙醇洗3次,抽滤,所得固体用水-乙醇-丙酮混合溶剂重结晶,5℃以下放置,使结晶充分洗出,抽滤,40℃真空干燥4h,得类白色结晶60.3g。hplc法检测纯度为99.08%;卡尔费休法测定水分为7.25%,热重分析失重为7.19%;元素分析理论值为:c:35.74%,h:3.58%,n:16.03%,na:5.26%,o:24.72%,s:14.68%;实测值为:c:35.71%,h:3.59%,n:16.05%,na:5.24%,o:24.73%,s:14.69%。试验例1:稳定性试验本发明人对本发明实施例1~3制备的1/2水头孢唑肟钠化合物和对比例1~2制备的头孢唑肟钠化合物进行了加速稳定性考察试验。考察条件为温度40℃±2℃,放置6个月,分别于0、1、2、3、6月末取样。考察指标为性状、澄清度、溶液颜色、水分、酸碱度、含量、头孢唑肟聚合物及有关物质。加速试验考察结果见下表。结论:由上述结果可知,通过6个月加速试验,本发明实施例1~3制备的样品各项检测指标明显优于对比例1~2的产品,充分说明了本发明制备的1/2水头孢唑肟钠化合物稳定性更好,质量优于同类产品,同时实施例1~3和对比例2的水分基本没有明显变化,推断其含有的水均为结晶水,而非吸附水;对比例1的水分明显下降,推断其含有的水为吸附水。试验例2:吸湿性试验将本发明实施例1~3制备的1/2水头孢唑肟钠化合物与对比例1~2制备的头孢唑肟钠化合物置于动态蒸汽吸附仪中40℃条件下,记录三小时内的重量变化,试验结果如下:相对湿度rh/%实施例1实施例2实施例3对比例1对比例200000010000002000.010003000.0200.020.034000.020.010.080.055000.020.020.250.18600.010.050.030.560.33700.670.580.600.980.87结论:本发明提供的1/2水头孢唑肟钠化合物更不易吸湿。试验例3:流动性试验本发明人对本发明实施例1~3和对比例1~2所制备的头孢唑肟钠采取漏斗法测定休止角,对头孢唑肟钠的流动性进行了研究。休止角检测结果:休止角检测结果:实施例休止角θ实施例128.2°实施例227.9°实施例327.6°对比例134.6°对比例235.2°结果:本发明制备的1/2水头孢唑肟钠化合物的流动性明显高于对比例1~2头孢唑肟钠化合物,可以满足不同制剂制备的需要。试验例4结晶水验证考察为了充分验证本发明头孢唑肟钠化合物中的1/2水为结晶水,本发明人通过热重分析法、60℃热稳定性10天、冷冻真空干燥失重法三种方法,考察各实施例和对比例的水分结果,具体如下:4、热重分析法热重分析是样品在高温状态下分解前的失重,是验证结晶水或吸附水的重要方法,本发明人分别对各实施例和对比例制备的头孢唑肟钠化合物进行了热重分析,结果汇总如下:实施例热重法失重(%)实施例12.19实施例22.16实施例32.20对比例14.40对比例27.19结果,实施例1~3制备的1/2水头孢唑肟钠化合物失重与含有的1/2个水的结果(理论值2.17%)基本一致;对比例1制备的头孢唑肟钠失重较大;对比例2制备的头孢唑肟钠化合物失重与含有的1.75个水的结果(理论值7.21%)基本一致。推断本发明实施例1~3和对比例2制备的头孢唑肟钠化合物所含水为结晶水,对比例1制备的头孢唑肟钠化合物所含水为吸附水。2、60℃热稳定性10天将本发明实施例制备的1/2水头孢唑肟钠化合物和对比例1~2制备的头孢唑肟钠化合物分别置于60℃烘箱中10天,分别于0、10天用卡尔费休法检测水分,结果如下:实施例0天(%)10天(%)实施例12.172.15实施例22.132.10实施例32.222.21对比例14.310.51对比例27.257.18结果,高温60℃放置10天,实施例1~3制备的1/2水头孢唑肟钠化合物水分基本没有明显变化,对比例1制备的头孢唑肟钠水分明显降低,对比例2制备的头孢唑肟钠化合物水分没有明显变化。推断本发明实施例1~3和对比例2制备的头孢唑肟钠化合物所含水为结晶水,对比例1制备的头孢唑肟钠化合物所含水为吸附水。3、冷冻真空干燥10小时将本发明实施例制备的1/2水头孢唑肟钠化合物和对比例1~2制备的头孢唑肟钠化合物分别置于-45℃冷冻干燥机中抽真空10小时,分别于0、10小时用卡尔费休法检测水分,结果如下:实施例0小时(%)10小时(%)实施例12.172.14实施例22.132.11实施例32.222.19对比例14.310.42对比例27.257.20结果,低温-45℃冷冻真空干燥10小时,实施例1~3制备的1/2水头孢唑肟钠化合物水分基本没有明显变化,对比例1制备的头孢唑肟钠水分明显降低,对比例2制备的头孢唑肟钠化合物水分没有明显变化。推断本发明实施例1~3和对比例2制备的头孢唑肟钠化合物所含水为结晶水,对比例1制备的头孢唑肟钠化合物所含水为吸附水。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1