一种3‑取代‑3‑羟基‑2‑吲哚酮类化合物的制备方法与流程
本发明涉及一种tmepo促进的3-取代-2-吲哚酮类化合物c(sp3)-h羟基化方法,尤其涉及一种以3-取代-2-吲哚酮衍生物为原料、在tempo、室温条件下发生羟基化反应,制备获得3-取代-3-羟基-2-吲哚酮类化合物的方法。
背景技术:
天然产物有多种多样的化学功能和性质,它们对药物的开发有重要作用。在这些物质当中,吲哚酮有着前所未有的结构多样性和生物活性,因此引起了化学家和生物学家广泛持久的关注。作为吲哚酮天然产物的一个子类,3-羟基-2-吲哚酮的骨架同样也是非常重要的生物单元。有越来越多的3-取代的3-羟基-2-吲哚酮结构被发现是构成很多的天然产物的核心骨架,并且具有广泛生物活性。其中比较有代表性的有如下几种(见下式一):
由于3-取代-3-羟基-2-吲哚酮类化合物及其衍生物是非常重要的有机中间体,以及许多天然化合物都含有这些骨架,其显示出具有良好的药物活性,在医药、生物等方面应用十分广泛。因此,发展便利、环境友好、高效地合成3-取代-3-羟基-2-吲哚酮及其衍生物的方法显得相当重要。
发明人发现,现有技术中合成制备3-取代-3-羟基-2-吲哚酮类化合物的合成途径主要包括如下两种:一是通过环合反应制备,另一种则是靛红的加成反应制备。近年来,也有科研工作者开发出了以3-取代-2-吲哚酮类化合物为原料制备3-取代-3-羟基-2-吲哚酮类化合物的方法。例如,可参见如下文献:
(1)“adinuclearpalladiumcatalystforr-hydroxylationofcarbonylswitho2”,tobiasritteret.al.,j.am.chem.soc.2011,133,1760-1762;
(2)“acatalytic,mildandefficientprotocolforthec-3aerialhydroxylationofoxindoles”,benjaminr.buckleyet.al.,tetrahedronletters54(2013)843-846;
(3)“ruthenium-catalyzeddirectα-alkylationofamidesusingalcohols”,boopathygnanaprakasamet.al.,org.biomol.chem.,2016,14,9215;
(4)“literaturesurveyandfurtherstudiesonthe3-alkylationofn-unprotected3-monosubstitutedoxindoles.practicalsynthesisofn-unprotected3,3-disubstitutedoxindolesandsubsequenttransformationsonthearomaticring”,balázsvolket.al.,molecules2017,22,24;
(5)“transition-metal-freec-hhydroxylationofcarbonylcompounds”,boopathygnanaprakasamet.al.,org.lett.2017,19,3628-3631;
(6)cn103613478a,20140305。
然而,在这些现有技术报道的3-取代-2-吲哚酮类化合物为原料制备3-取代-3-羟基-2-吲哚酮类化合物方法中,其反应条件较为苛刻,例如使用-78℃的低温,使用丁基锂、叔丁醇钾等强碱,和/或使用贵金属钯、钌、磷配体等价格昂贵的催化体系等。因此,寻找一种更高效、更廉价、更绿色的合成方法仍然是一个挑战性的课题。
技术实现要素:
本发明目的在于克服现有技术的不足,提供一种工艺简单、高效廉价的制备3-取代-3-羟基-2-吲哚酮类化合物的绿色合成方法,该方法以3-取代-2-吲哚酮类化合物为原料,在室温及tempo促进下,方便地并以优异的产率制备获得3-取代-3-羟基-2-吲哚酮类化合物。
本发明提供的3-取代-3-羟基-2-吲哚酮类化合物的制备方法,该方法以3-取代-2-吲哚酮类化合物为原料,通过下列步骤进行制备获得:
向schlenk反应瓶中加入3-取代-2-吲哚酮衍生物(1a)、式2a的化合物和有机溶剂,将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全,经后处理得到目标产物3-取代-3-羟基-2-吲哚酮类化合物(i)。
本发明提供的3-取代-3-羟基-2-吲哚酮类化合物的制备方法,其化学反应式可表述为(见式二):
上述的反应中,所述有机溶剂可选自四氢呋喃、1,4-二氧六环、二氯甲烷、乙腈中的一种或几种,优选为上述溶剂的无水溶剂,最优选为无水四氢呋喃。
上述的反应中,所述的反应气氛为1atm的空气气氛,也可以替换为1atm的氧气气氛,在氧气气氛下收率略高于在空气条件下,从经济成本等方面考虑,优选为空气气氛。
所述的后处理操作如下:将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物3-取代-3-羟基-2-吲哚酮类化合物(i)。
上述式1a表示的原料3-取代-2-吲哚酮类化合物及式i表示的产物3-取代-3-羟基-2-吲哚酮类化合物中,r1表示其所连接的苯环上1个或多个取代基,各个r1彼此独立地选自氢、c1-c6烷基、c1-c6烷氧基、c1-c6酰基、c1-c6酯基、卤素、氰基、硝基、c3-c6环烷基、c5-c14芳基、c5-c14杂芳基、-nrarb。其中,ra,rb彼此独立地选自c1-c6烷基或氢;所述杂芳基的杂原子选自o、s或n。
r2表示氢、c1-c6烷基、卤素、氰基、c3-c6环烷基、c5-c14芳基、c5-c14杂芳基;所述杂芳基的杂原子选自o、s或n。
r3表示氢、叔丁氧羰基(boc)、c1-c6烷基、c1-c6酰基、c3-c6环烷基、c5-c14芳基、c5-c14芳基-c1-c6烷基、c5-c14杂芳基,所述杂芳基的杂原子选自o、s或n。
其中,上述各烷基、烷氧基、环烷基、芳基和杂芳基可以进一步地被取代基取代,所述的取代基选自卤素或c1-c6的烷基。
优选地、r1表示其所连接的苯环上1个或多个取代基,各个r1彼此独立地选自氢、c1-c6烷基、卤素、c1-c6烷氧基、硝基、c5-c14芳基;其中所述c1-c6烷基和/或c5-c14芳基可以进一步被取代,所述取代基选自卤素或c1-c6的烷基。
优选地,r2表示氢、c1-c6烷基、卤素、氰基、c5-c14芳基,其中所述c1-c6烷基和/或c5-c14芳基可以进一步被取代,所述取代基选自卤素或c1-c6的烷基。
优选地,r3表示氢、叔丁氧羰基(boc)、c1-c6烷基、c1-c6酰基、苯基、苄基,其中所述c1-c6烷基和/或c1-c6酰基、苯基和/或苄基可以进一步被取代,所述取代基选自卤素或c1-c6的烷基。
上述式2a表示的化合物中,所述r4表示氢,-oh,-oac;当r4表示氢时,该化合物即本领域所熟知的tempo。
其中,上述的以及本说明书中其它部分所述的tempo属于本领域公知的化学物质缩写形式,其为2,2,6,6-四甲基哌啶-氮-氧化物,英文名称为2,2,6,6-tetramethylpiperidine1-oxyl。tempo或其它式2a的化合物的加入量优选为3-取代-2-吲哚酮衍生物(1a)加入量的10~40mol%,最优选为3-取代-2-吲哚酮衍生物(1a)加入量的20mol%。
本发明的有益效果是:提出了一种制备3-取代-3-羟基-2-吲哚酮类化合物的新方法,该方法以3-取代-2-吲哚酮类化合物为反应原料,在室温、和tempo促进下发生反应c(sp3)-h羟基化反应,以高收率得到一系列的目标产物。该方法不使用碱、不使用金属催化剂、反应温度为室温、反应气氛为空气,具有反应底物适应范围广泛、简单高效、经济环保的优点,特别适合于工业化生产。
附图说明
图1为产物3-羟基-3-苯基-2-吲哚酮的核磁氢谱图。
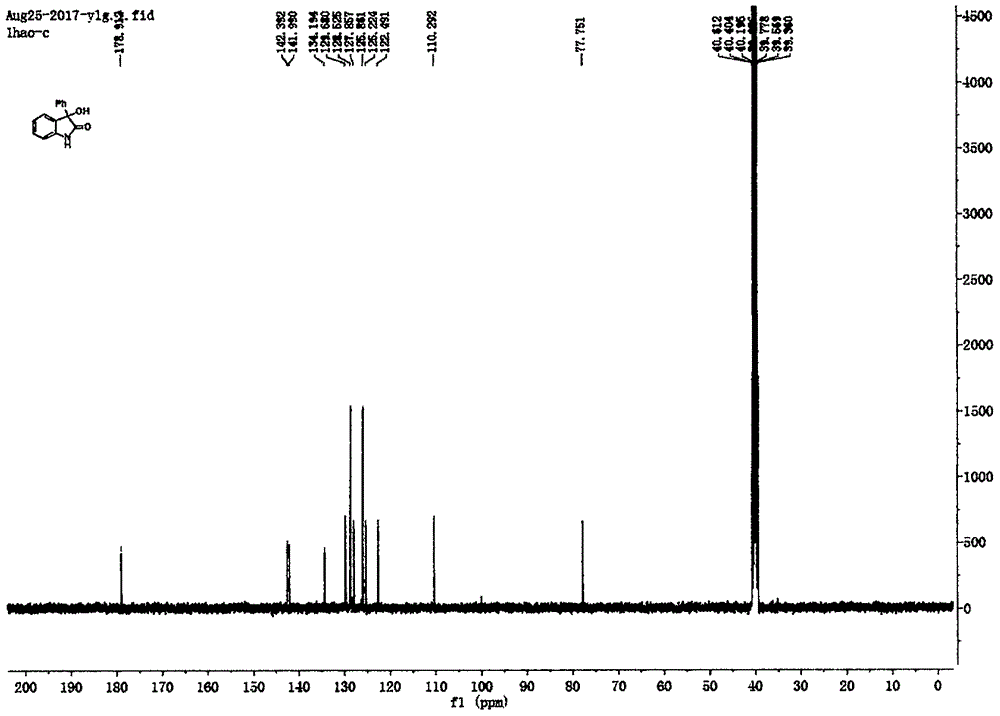
图2为产物3-羟基-3-苯基-2-吲哚酮的核磁碳谱图。
图3为本发明反应的机理示意图。
具体实施方式
以下结合具体实施例,对本发明进行进一步详细的描述。
实施例1化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物3-苯基-3-羟基-2-吲哚酮。
白色固体(0.0628g,93%yield);1hnmr(400mhz,dmso-d6)δ:10.39(s,1h),7.33-7.23(m,6h),7.09(d,j=7.2,1h),6.96(t,j=7.6hz,1h),6.90(d,j=7.6hz,1h),6.61(s,1h);13cnmr(100mhz,dmso-d6)δ:178.9,142.4,142.0,134.2,129.7,128.5,127.9,125.9,125.2,122.5,110.3,77.8;hrmsm/z(esi)calcdforc14h11no2na([m+na]+)248.0682,found248.0680。
实施例2用溶剂乙腈替换无水四氢呋喃,其余条件同实施例1,目标产物收率为79%。
实施例3用溶剂二氯甲烷替换无水四氢呋喃,其余条件同实施例1,目标产物收率为70%。
实施例4用溶剂1,4-二氧六环替换无水四氢呋喃,其余条件同实施例1,目标产物收率为86%。
实施例5用溶剂甲苯替换无水四氢呋喃,其余条件同实施例1,目标产物收率为0%。
实施例6调整tempo的加入量为10mmol%,即0.03mmol,其余条件同实施例1,目标产物收率为74%。
实施例7调整tempo的加入量为40mmol%,即0.12mmol,其余条件同实施例1,目标产物收率为93%。
实施例8调整反应气氛为氧气气氛(1atm),其余条件同实施例1,目标产物收率为95%。
实施例9调整反应气氛为氮气气氛(1atm),其余条件同实施例1,目标产物收率为0%。
实施例10化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的6-甲氧基-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0620g,81%yield);1hnmr(400mhz,dmso-d6)δ:10.27(s,1h),7.32-7.22(m,6h),6.83(d,j=7.2hz,1h),6.74(d,j=8.4hz,1h),5.81(s,1h),3.34(s,3h);13cnmr(100mhz,dmso-d6)δ:175.6,153.4,136.0,130.8,129.8,127.6,126.7,125.4,114.1,113.6,109.9,77.1,55.1;hrmsm/z(esi)calcdforc15h13no3na([m+na]+)278.0788,found278.0792。
实施例11化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的6-氯-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0707g,91%yield);1hnmr(400mhz,dmso-d6)δ:10.56(s,1h),7.35-7.27(m,6h),7.10(s,1h),6.92(d,j=8.0hz,1h),6.78(s,1h);13cnmr(100mhz,dmso-d6)δ:178.5,141.3,136.2,129.6,128.7,128.2,128.1,126.4,125.8,125.2,111.9,77.8;hrmsm/z(esi)calcdforc14h10clno2na([m+na]+)282.0292,found282.0294。
实施例12化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的6-溴-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0839g,92%yield);1hnmr(400mhz,dmso-d6)δ:10.57(s,1h),7.44(d,j=8.0hz,1h),7.35-7.27(m,5h),7.21(s,1h),6.88(d,j=8.0hz,1h),6.78(s,1h);13cnmr(100mhz,dmso-d6)δ:178.4,141.7,141.3,136.6,132.4,128.7,128.1,127.8,125.8,114.1,112.5,77.8;hrmsm/z(esi)calcdforc14h10brno2na([m+na]+)325.9787,found325.9785。
实施例13化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的6-硝基-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0688g,85%yield);1hnmr(400mhz,dmso-d6)δ:11.16(s,1h),8.25(d,j=10.4hz,1h),7.90(s,1h),7.36-7.30(m,5h),7.13(d,j=8.8hz,1h),7.00(s,1h);13cnmr(100mhz,dmso-d6)δ:179.0,148.9,142.9,140.5,135.0,128.9,128.5,127.1,125.8,120.6,110.8,77.3;hrmsm/z(esi)calcdforc14h10n2o4na([m+na]+)293.0533,found293.0529。
实施例14化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的3-(2,5-二甲基)苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0684g,90%yield);1hnmr(400mhz,dmso-d6)δ:10.51(s,1h),7.70(s,1h),7.23(t,j=7.6hz,1h),6.99(d,j=7.6hz,1h),6.92-6.87(m,3h),6.82(d,j=7.2hz,1h),6.54(s,1h),2.33(s,3h),1.72(s,3h);13cnmr(100mhz,dmso-d6)δ:178.2,143.0,139.3,134.6,132.9,131.5,131.3,129.8,128.4,127.7,124.8,122.4,110.1,77.0,21.4,18.7;hrmsm/z(esi)calcdforc16h15no2na([m+na]+)276.0995,found276.0997。
实施例15化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的3-乙基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
黄色固体(0.0436g,82%yield);1hnmr(400mhz,dmso-d6)δ:10.20(s,1h),7.25-7.18(m,2h),6.96(t,j=7.6hz,1h),6.80(d,j=7.6hz,1h),5.80(s,1h),1.79-1.73(m,2h),0.61(t,j=7.6hz,3h);13cnmr(100mhz,dmso-d6)δ:179.8,142.3,132.3,129.2,124.3,122.0,109.9,76.6,31.1,7.9;hrmsm/z(esi)calcdforc10h11no2na([m+na]+)200.0682,found200.0684。
实施例16化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的n-甲基-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0374g,52%yield);1hnmr(400mhz,dmso-d6)δ:7.38-7.26(m,6h),7.14(d,j=7.2hz,1h),7.10-7.03(m,2h),6.68(s,1h),3.16(s,3h);13cnmr(100mhz,dmso-d6)δ:177.1,143.9,141.7,133.5,129.8,128.6,128.0,125.9,124.8,123.2,109.3,77.5,26.6;hrmsm/z(esi)calcdforc15h13no2na([m+na]+)262.0838,found262.0840。
实施例17化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的n-苄基-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0577g,61%yield);1hnmr(400mhz,dmso-d6)δ:7.36-7.24(m,11h),7.17(d,j=7.2hz,1h),7.03(t,j=7.6hz,1h),6.97(d,j=8.0hz,1h),6.85(s,1h),4.91(s,2h);13cnmr(100mhz,dmso-d6)δ:177.3,142.9,141.8,136.8,133.5,129.7,129.1,128.6,128.1,127.9,127.7,125.9,125.0,123.3,109.9,77.5,43.2;hrmsm/z(esi)calcdforc21h17no2na([m+na]+)338.1151,found338.1149。
实施例18化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的n-苯基-3-苯基-2-吲哚酮、0.06mmol的tempo和无水四氢呋喃(2ml),将反应瓶置于室温、空气气氛条件下搅拌反应,经tlc或gc监测反应进程,至原料反应完全(约18小时),将反应完成后的反应液减压浓缩,将残余物经柱层析分离(洗脱溶剂为:乙酸乙酯/正己烷)得到目标产物。
白色固体(0.0569g,63%yield);1hnmr(400mhz,dmso-d6)δ:7.60(t,j=7.6hz,2h),7.48(t,j=8.4hz,3h),7.41-7.34(m,4h),7.30(t,j=7.6hz,2h),7.24(d,j=7.6hz,1h),7.11(t,j=7.6hz,1h),6.92(s,1h),6.81(d,j=8.0hz,1h);13cnmr(100mhz,dmso-d6)δ:176.7,143.5,141.6,134.6,133.5,130.2,129.9,128.7,128.6,128.1,127.1,125.9,125.4,123.9,109.7,77.5;hrmsm/z(esi)calcdforc20h15no2na([m+na]+)324.0995,found324.0991.
实施例19用tempo结构类似物替换tempo合成化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的3-苯基-2-吲哚酮、0.06mmol的
实施例20用tempo结构类似物替换tempo合成化合物
向容积为20ml的schlenk反应瓶中加入0.3mmol的3-苯基-2-吲哚酮、0.06mmol的
- 还没有人留言评论。精彩留言会获得点赞!