2,7‑二溴‑9,9‑二烷基‑1,6‑二硝基‑芴及其制备方法与流程
本发明涉及芴类衍生物的制备方法,具体是一种2,7-二溴-9,9-二烷基-1,6-二硝基-芴及其制备方法,属于有机化学合成领域。
背景技术:
有机电致发光材料的研究和开发一直是科学研究领域的焦点,稳定和高效的红、绿、蓝三基色发光材料是实现有机电致发光器件全色显示的关键。如今红色和绿色发光材料的研发已经趋近成熟,其发光效率和寿命已经达到了产业化的要求,然而蓝光材料还存在着稳定性差、效率低和寿命短等问题,需要不断的研究改进,使oleds器件更好的实现全色彩显示以及推进白光器件的产业化步伐。因此,在oleds的研究中,蓝光材料起着至关重要的作用。蓝光材料不仅要有宽带隙、匹配的电子亲和势(ea)和第一电离能(ip),而且还要具有大的共轭吸收波长和小的分子偶极矩。
在各种有机电致发光材料中,芴是一种具有特殊联苯结构的化合物,其结构有利于在其苯环的几个反应点上引入不同的基团(卤素原子、烷基、硝基、芳基、酯基等)得到一系列衍生物,实现不同的光电性质。因此芴及其衍生物具备宽能隙(大于2.90ev)、易修饰、大的共轭吸收波长、高荧光量子效率(60%~80%)、高光热稳定性和发光效率、良好的光电学性质等多种特性,被广泛应用在有机电致发光二极管、有机场效应晶体管、有机太阳能电池、化学和生物传感器等方面。特别是在有机电致发光器件领域极具开发前景,芴及其衍生物成为了一种备受青睐的蓝光材料。
tzu-chaulin等人在芴的3,6位上引入两个硝基,得到2,7-二乙酰胺基-9,9-二己基-3,6-二硝基-芴。xichuanli等人也在芴的3,6位上引入两个硝基,得到2,7-二甲磺酰胺基-9,9-二异辛基-3,6-二硝基-芴。而本发明所得到的是特殊的不对称1,6位双硝化芴,其具备了一定的空间位阻和分子扭转张力,不易造成分子间的π-π堆叠,可以有效防止激基缔合物的形成,从而提高光谱强度、稳定性和效率。
技术实现要素:
本发明所解决的技术问题是提供一种2,7-二溴-9,9-二烷基-1,6-二硝基-芴及其制备方法,其特征在于所述的2,7-二溴-9,9-二烷基-1,6-二硝基-芴具有如下通式:
一种如前述2,7-二溴-9,9-二烷基-1,6-二硝基-芴的制备方法,其特征在于该制备方法包括以下步骤:将芴溶于氯仿溶液,将溶解br2的氯仿溶液滴入上述溶液中,0℃避光搅拌10小时,停止反应将混合溶液倒入na2s2o3水溶液中,用二氯甲烷萃取,合并有机层分别用去离子水和饱和食盐水洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物在无水乙醇溶液中重结晶,得到白色固体2,7-二溴-芴;然后,将上一步反应所得2,7-二溴-芴与四丁基溴化铵和卤代烃溶于甲苯和50%质量分数的naoh水溶液中,氩气保护80℃回流搅拌24小时,停止反应用乙酸乙酯萃取溶液,合并有机层分别用去离子水和饱和食盐水洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离,得到淡黄色固体2,7-二溴-9,9-二烷基-芴;最后,将上一步反应所得到的2,7-二溴-9,9-二烷基-芴,溶解于二氯甲烷溶液中,加入浓硫酸,冷却至0℃,硝酸铈铵分批加入上述混合溶液,搅拌反应1小时,停止反应加入去离子水,用二氯甲烷萃取,有机相分别用去离子水和饱和食盐水洗涤,加入无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离,得到橙黄色的产物2,7-二溴-9,9-二烷基-1,6-二硝基-芴。前面所述的2,7-二溴-9,9-二烷基-1,6-二硝基-芴的制备方法,其特征在于,2,7-二溴-9,9-二烷基-芴与硝酸铈铵的物料摩尔比为1:3,反应溶剂为二氯甲烷,反应温度为0℃,反应时间为1小时。
本发明设计和合成了一类未见报道的不对称双硝化芴:2,7-二溴-9,9-二烷基-1,6-二硝基-芴。它们特殊的不对称双硝化结构使目标产物具备了一定的空间位阻和分子扭转张力,不易造成分子间的π-π堆叠,可以有效防止激基缔合物的形成,从而提高光谱质量、稳定性和效率。这种不对称双硝化结构也可能产生一定的非线性光学特性,调节芴类化合物的光电性能,一定程度上改善了芴结构的缺点,为研究合成新型光电功能材料提供了发展的空间。此外,本发明在芴的硝化方面首次采用硝酸铈铵,反应条件较为温和快速,产率高达70%以上。
附图说明
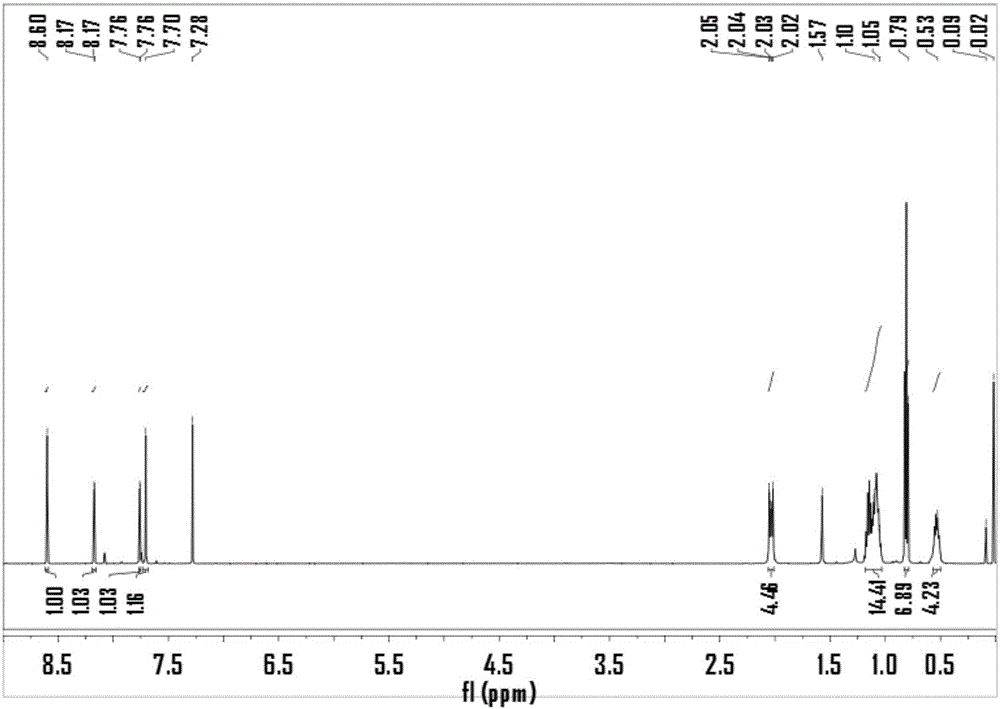
图1:2,7-二溴-9,9-二己基-1,6-二硝基-9h-芴的1hnmr谱图。
图2:2,7-二溴-9,9-二己基-1,6-二硝基-9h-芴的13cnmr谱图。
图3:2,7-二溴-9,9-二己基-1,6-二硝基-9h-芴的1h-1hcosy谱图。
图4:2,7-二溴-9,9-二己基-1,6-二硝基-9h-芴在ch2cl2溶液中的紫外-可见吸收光谱图。
具体实施方式
为对本发明进行更好的说明,列举实施例如下:
实施例1:
2,7-二溴-9,9-二十二烷基-1,6-二硝基-9h-芴的合成
将芴(5.00g,30.08mmol)溶于40ml氯仿溶液,将溶解br2(3.08ml)的15ml氯仿溶液滴入上述溶液中,0℃避光搅拌10小时,停止反应将混合溶液倒入na2s2o3水溶液中,用二氯甲烷萃取(15ml×3),合并有机层分别用去离子水(25ml×3)和饱和食盐水(20ml)洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物在无水乙醇溶液中重结晶,得到白色固体2,7-二溴-芴(7.04g,72.2%);然后,将上一步反应所得2,7-二溴-芴(6.40g,19.75mmol)与四丁基溴化铵(0.10g,0.31mmol)和1-溴十二烷(10ml,41.65mmol)溶于60ml甲苯和25ml50%质量分数的naoh水溶液中,氩气保护80℃回流搅拌24小时,停止反应用乙酸乙酯(25ml×3)萃取溶液,合并有机层分别用去离子水(25ml×3)和饱和食盐水(30ml)洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离(石油醚:二氯甲烷=4:1),得到淡黄色固体2,7-二溴-9,9-二十二烷基-芴(11.30g,86.6%);最后,将上一步反应所得到的2,7-二溴-9,9-二十二烷基-芴(2.00g,3.03mmol),溶解于60ml的二氯甲烷溶液中,加入10ml的浓硫酸,冷却至0℃,硝酸铈铵(4.98g,9.08mmol)分批加入上述混合溶液,搅拌反应1小时,停止反应加入50ml去离子水,用二氯甲烷(15ml×3)萃取,有机相分别用去离子水(25ml×3)和饱和食盐水(20ml)洗涤,加入无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离(石油醚:二氯甲烷=4:1),得到橙黄色的产物2,7-二溴-9,9-二十二烷基-1,6-二硝基-芴(1.7g,74.56%)。1hnmr(500mhz,cdcl3)δ8.60(s,1h),8.17(d,j=1.7hz,1h),7.75(d,j=1.7hz,1h),7.70(s,1h),2.03(m,4h),1.32-1.15(m,36h),0.88(t,j=7.0hz,6h),0.58-0.50(m,4h)。13cnmr(126mhz,cdcl3)δ156.44,155.83,149.64,145.06,136.30,130.85,130.52,128.71,127.19,122.59,122.32,115.52,55.78,40.21,31.86,29.60,29.56,29.55,29.48,29.41,29.28,29.04,23.55,22.64,14.06。
实施例2:
2,7-二溴-9,9-二己基-1,6-二硝基-9h-芴的合成
将芴(5.00g,30.08mmol)溶于40ml氯仿溶液,将溶解br2(3.08ml)的15ml氯仿溶液滴入上述溶液中,0℃避光搅拌10小时,停止反应将混合溶液倒入na2s2o3水溶液中,用二氯甲烷萃取(15ml×3),合并有机层分别用去离子水(25ml×3)和饱和食盐水(20ml)洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物在无水乙醇溶液中重结晶,得到白色固体2,7-二溴-芴(7.04g,72.2%);然后,将上一步反应所得2,7-二溴-芴(6.40g,19.75mmol)与四丁基溴化铵(0.10g,0.31mmol)和1-溴己烷(7.30ml,44.24mmol)溶于60ml甲苯和25ml50%质量分数的naoh水溶液中,氩气保护80℃回流搅拌24小时,停止反应用乙酸乙酯(25ml×3)萃取溶液,合并有机层分别用去离子水(25ml×3)和饱和食盐水(30ml)洗涤,之后用无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离(石油醚:二氯甲烷=4:1),得到淡黄色固体2,7-二溴-9,9-二己基-芴(8.20g,84.36%);最后,将上一步反应所得到的2,7-二溴-9,9-二己基-芴(1.00g,2.03mmol),溶解于30ml的二氯甲烷溶液中,加入5ml的浓硫酸,冷却至0℃,硝酸铈铵(3.34g,6.09mmol)分批加入上述混合溶液,搅拌反应1小时。停止反应加入30ml去离子水,用二氯甲烷(15ml×3)萃取,有机相分别用去离子水(15ml×3)和饱和食盐水(20ml)洗涤,加入无水硫酸镁干燥有机层,用旋蒸仪除去溶剂,将粗产物用层析色谱柱分离(石油醚:二氯甲烷=2:1),得到橙黄色的产物2,7-二溴-9,9-二己基-1,6-二硝基-芴(0.62g,52.54%),图1为其1hnmr谱图,图2为其13cnmr谱图,图3为其1h-1hcosy谱图,图4为其在ch2cl2溶液中的紫外-可见吸收光谱图。1hnmr(500mhz,cdcl3)δ=8.60(s,1h),8.17(d,j=1.7,1h),7.76(d,j=1.7,1h),7.70(s,1h),2.03(m,4h),1.18-1.04(m,12h),0.81(t,j=7.2,6h),0.53(m,4h)。13cnmr(126mhz,cdcl3)δ=156.47,155.82,149.62,145.07,136.31,130.87,130.53,128.71,127.24,122.63,122.36,115.58,55.79,40.28,31.29,29.32,23.57,22.49,13.93。
显然,上述实施例的列举仅仅是为清晰的说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其他不同形式的变化或者改动。这里无需也无法对所有的实施方式给予穷举。而由此所引伸出的显而易见的变化或者改动仍然处于本发明创造的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!