聚集诱导发光探针及其制备与在钙离子检测和成像中的应用的制作方法
本发明属于钙离子检测领域,特别涉及一种聚集诱导发光探针及其制备方法与在钙离子检测和成像中的应用,所述应用是基于荧光信号变化检测钙离子。
背景技术:
钙是机体的重要组成元素之一,同时钙离子作为细胞内重要信使,调控多种生理功能,在生命活动中扮演着重要角色。因此,钙离子测定在生物医学研究中有着重要意义。例如,高血钙症是指血清中总钙和游离钙离子浓度的异常升高(总钙大于2.5mmol/l,游离钙大于1.4mmol/l),而引起高血钙症的病因有甲状旁腺功能亢进、代谢性酸中毒、维生素d过多症以及多种恶性肿瘤,包括乳腺癌、骨肿瘤、肺癌、胃癌、卵巢癌等。
另外,骨由羟基磷灰石基质构成,当其形成微小裂缝时,钙离子在微小裂缝区域会形成高浓度聚集,因此钙离子检测探针可有效实现对骨的微小裂缝区域的识别和成像,有助于对骨裂缝相关疾病的及早诊断和治疗。
钙盐可在各种软组织(脑、乳腺、血管、软骨、心脏瓣膜等)中沉积,并使其硬化形成钙化斑块。钙化斑块的识别和检测,有助于对脑膜瘤、乳腺癌、动脉粥状硬化等疾病的病理分析。当前临床上通常利用茜素红s染色钙化斑块,具有操作复杂、信噪比低、特异性差等缺陷,因此迫切需要发展操作简便、信噪比高的方法,实现对钙化斑块的特异性染色。
目前检测钙离子的方法主要有:核磁共振法、微电极法、比色法、原子吸收光谱法、离子色谱法和电化学分析法等。然而,这些方法存在着以下缺点:需要使用大型固定仪器、操作繁琐、成本高昂、难以直接测定游离钙离子的浓度等,不利于在基层医院推广使用。为了使钙离子的检测易于实施,迫切需要发展操作简便、成本低廉的有效检测方法。
近年来,荧光法检测钙离子表现出显著的优点,如:灵敏度高、操作简单、成本低等,在生命科学研究和临床检测领域,日益获得广泛的应用。然而,具有聚集猝灭发光(acq)性质的荧光材料在聚集态下,容易发生荧光的自猝灭,因此其针对钙离子的检测范围有限,而且其自身在溶液态具有高背景噪音,识别钙离子后的信噪比较低。
与聚集猝灭发光(acq)相反,具有聚集诱导发光(aie)性质的荧光探针,在稀溶液中发光很弱,但在聚集态发光很强。通过形成发光聚集体,aie探针作为新一代荧光探针,具有新颖的点亮检测机制、聚集态高发光效率、强抗光漂白能力、大的斯托克位移和低细胞毒性等优点,可有效克服聚集猝灭发光的缺陷,在分析检测和生物成像等领域日益获得广泛应用。具有聚集诱导发光(aie)性质的钙离子检测探针具有广阔的应用前景,不仅可以用于游离钙离子的检测,还特别适合于对钙离子聚集区域的特异性成像,包括骨的微小裂缝区域和软组织的钙化斑块成像等。
技术实现要素:
为了克服现有技术的缺点和不足,也为了实现对钙离子的高效检测,本发明的目的在于提供一种聚集诱导发光探针。所述探针具有aie性质,在水溶液中,由于分子内运动,不发出荧光,与钙离子发生鳌合作用后,形成聚集体,由于分子内运动受限,发出强荧光。
本发明聚集诱导发光探针的结构中含有分子内六元环氢键和可在溶解态自由旋转的n–n单键。在溶解态,由于分子内运动,不发出荧光;而在聚集态,分子间的紧密排列会抑制分子内n–n单键的旋转,而且会保护分子内氢键免受外界环境的干扰,有利于激发态分子内质子转移过程发生,从而发出强荧光。由于本发明的聚集诱导发光探针(式i化合物)具有很好的水溶性,而其荧光发射对分子的聚集态非常敏感,因此特别适于通过生成聚集体,实现对生理环境下钙离子的检测。
本发明的另一目的在于提供上述聚集诱导发光探针的制备方法。
本发明的再一目的在于提供上述聚集诱导发光探针在钙离子检测和成像中的应用。所述聚集诱导发光探针在含有钙离子的溶液中,可通过在紫外光照射下观察或检测荧光变化。
本发明的聚集诱导发光探针(式i化合物)在水溶液中,具有很好的溶解性,几乎不发出荧光,但当其与钙离子结合生成聚集体后,会发出很强的荧光,因此可用作优异的点亮探针,实现对钙离子的宽范围定量检测。
本发明目的基于如下技术方案实现:
一种聚集诱导发光探针,其结构为式i:
其中:
r为氢、烷基、全氟烷基、烷基氧基(烷氧基结构式为r1-o-,r1为烷基)、烷基巯基(结构式为r2-s-,r2为烷基)、烷基胺基(结构式为r3-nh-,r,3为烷基)、卤素、氰基、硝基、芳基、杂芳基;
x为直键(直键是指x不存在)、亚烷基;y为直键(直键是指y不存在)、亚芳基、亚杂芳基;
a+为阳离子,优选为金属离子;
所述烷基为直链或支链烷基;优选为c1-10的烷基(碳数为1~10的烷基),进一步优选为c1-6的烷基,更优选为c1-3的烷基;
所述烷基氧基中的烷基、烷基巯基中烷基、烷基胺基中烷基各自独立为直链或支链烷基,优选为c1-10的烷基(碳数为1~10的烷基),进一步优选为c1-6的烷基,更优选为c1-3的烷基;
所述芳基为具有6-20个碳原子的单环或多环芳族基团(具有6-20个碳原子的单环或多环芳族化合物失去一个氢形成的基团),优选为苯基、萘基、蒽基或芘基;
所述杂芳基为具有1-20个碳原子、1-4个选自n、s、o杂原子的单环或多环杂芳族基团(即单环或多环杂芳族化合物失去一个氢形成的基团);且当碳原子数为1时,杂原子数≥2,当杂原子数为1时,碳原子数≥2;所述杂芳基优选为吡咯基、吡啶基、嘧啶基、咪唑基、噻唑基、吲哚基、氮杂萘基、氮杂蒽基或氮杂芘基(即吡咯、吡啶、嘧啶、咪唑、噻唑、吲哚、氮杂萘、氮杂蒽、氮杂芘各自失去一个氢形成的基团);
所述亚烷基为直链或支链亚烷基;优选为c1-10的亚烷基(碳数为1~10的亚烷基),进一步优选为c1-6的亚烷基,更优选为c1-3的亚烷基;
所述亚芳基为具有6-20个碳原子的单环或多环芳族基团(即单环或多环芳族化合物失去两个氢形成的基团),优选为亚苯基、亚萘基、亚蒽基或亚芘基(即苯、萘、蒽、芘各自失去两个氢形成的基团),更优选为1,4-对苯基,1.5-萘基;
所述亚杂芳基为具有1-20个碳原子、1-4个选自n、s、o杂原子的单环或多环杂芳族基团(即单环或多环杂芳族化合物失去两个氢形成的基团);且当碳原子数为1时,杂原子数≥2,当杂原子数为1时,碳原子数≥2;所述亚杂芳基优选为亚吡咯基、亚吡啶基、亚嘧啶基、亚咪唑基、亚噻唑基、亚吲哚基、亚氮杂萘基、亚氮杂蒽基或亚氮杂芘基(即吡咯、吡啶、嘧啶、咪唑、噻唑、吲哚、氮杂萘、氮杂蒽、氮杂芘各自失去两个氢形成的基团);
所述阳离子为钠离子、钾离子、铷离子或铯离子。
优选地,所述聚集诱导发光探针的结构式式i中:
r为氢、x为亚甲基、y为直键、a+为钠离子(na+)。
所述聚集诱导发光探针(式i化合物)的制备方法,包括以下步骤:
(1)在有机溶剂中以及弱酸性环境下,式iv化合物与式v化合物反应,得到式iii化合物;
(2)在有机溶剂中,式iii化合物在碱性溶液的作用下发生水解反应,得到式ii化合物;
(3)在有机溶剂中,式ii化合物与醇盐反应,得到式i化合物即聚集诱导发光探针;
式ii化合物为
步骤(3)中所述有机溶剂为醇类物质,优选为c1-c6的一元醇,例如甲醇、乙醇、丁醇、叔丁醇,更优选甲醇。
所述醇盐为乙醇金属盐、甲醇金属盐,优选为甲醇钠、乙醇钠、甲醇钾、乙醇钾,甲醇铷、乙醇铷、甲醇铯、乙醇铯,更优选为甲醇钠。
步骤(3)中所述反应的温度为20~80℃,反应的时间为0.1~0.5h;式ii化合物与醇盐的摩尔比为1:4;
步骤(3)的反应方程式如下:
其中,r、x、y、a+如上述所定义。
步骤(2)中所述有机溶剂为四氢呋喃、二甲基亚砜、n,n-二甲基甲酰胺,优选为四氢呋喃。
步骤(2)中所述碱性溶液为氢氧化钾水溶液、氢氧化钠水溶液,优选为氢氧化钠水溶液。所述碱性溶液的浓度为0.1mol/l-10.0mol/l。
步骤(2)中所述碱性溶液中碱性化合物与式iii化合物的摩尔比为4:1;所述反应的温度为20~80℃,反应的时间为1~4h;
步骤(2)中反应的方程式如下:
其中,r、x、y、a+如上述所定义。
步骤(1)中所述有机溶剂为乙醇、乙腈、四氢呋喃、二甲基亚砜、n,n-二甲基甲酰胺,优选为乙醇。
步骤(1)中所述弱酸性环境是指加入弱酸,所述弱酸为醋酸、甲酸、三氟乙酸,优选为醋酸。
所述式iv化合物与弱酸的用量比为1mmol:(40~200)μl。
步骤(1)中所述式iv化合物与式v化合物的摩尔比为(2~3):1;所述反应的温度为20~80℃,反应的时间为15min~2h;
步骤(1)中反应的方程式如下:
其中,r、x、y、a+如上述所定义。
上述聚集诱导发光探针在钙离子检测和成像中的应用。
本发明的聚集诱导发光探针(式i化合物)在水溶液中具有很好的溶解性,当其以单分子态分散于水溶液中时,由于分子内运动,几乎不发出荧光,但当其与钙离子通过鳌合作用形成聚集体时,由于分子内运动受到限且分子内氢键可免受外界环境的干扰,可发出强荧光,因此可以通过荧光强度变化确定水溶液中钙离子的浓度。
本发明聚集诱导发光探针(式i化合物)表现出非常低的细胞毒性,有利于其生物成像和检测应用。
与现有技术相比,本发明具有以下有益效果:
1、本发明聚集诱导发光探针(式i化合物)对钙离子检测,具有宽响应范围,有望用于对高钙血症的检测;
2、本发明聚集诱导发光探针(式i化合物)对钙离子的检测机制新颖,通过生成聚集体发出强荧光,点亮比率高达10倍以上,具有高信噪比的特点,有效克服传统荧光材料的聚集猝灭发光的缺陷;
3、本发明聚集诱导发光探针(式i化合物)作为钙离子检测探针,具有制备简易、成本低、操作方便、检测范围广等优点,有望用于偏远地区或基层临床上的钙离子检测;
4、本发明聚集诱导发光探针(式i化合物)特别适于对钙离子高浓度聚集区域的特异性成像和检测,包括骨的微小裂缝和软组织的钙化斑块等,检测效果好。
附图说明
图1为实施例1制备的式iii-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式iii-1化合物在四氢呋喃溶液中的归一化紫外吸收光谱图;(b)为式iii-1化合物在四氢呋喃和水的混合溶液中不断增加水含量的荧光发射光谱图;(c)为式iii-1化合物在四氢呋喃和水的混合溶液中不断增加水含量的最大荧光发射强度与四氢呋喃溶液中最大荧光发射强度的比值变化图;λex=366nm;
图2为实施例1制备的式ii-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式ii-1化合物在二甲基亚砜溶液中的归一化紫外吸收光谱图;(b)为式ii-1化合物在二甲基亚砜和乙醇的混合溶液中不断增加乙醇含量的荧光发射光谱图;(c)为式ii-1化合物在二甲基亚砜和乙醇的混合溶液中不断增加乙醇含量的最大荧光发射强度与二甲基亚砜溶液中最大荧光发射强度的比值变化图;λex=363nm;
图3为实施例1制备的式i-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式i-1化合物在水溶液中的归一化紫外吸收光谱图;(b)为式i-1化合物在水和四氢呋喃的混合溶液中不断增加四氢呋喃含量的荧光发射光谱图;(c)为式i-1化合物在四氢呋喃和水的混合溶液中不断增加四氢呋喃含量的最大荧光发射强度与水溶液中最大荧光发射强度的比值变化图;λex=351nm;
图4为实施例1制备的式i-1化合物在不同ph的水溶液(ph=4.0,6.0,7.0,7.4)的归一化紫外吸收图;
图5为钙离子浓度对实施例1制备的式i-1化合物荧光性能的影响图;(a)为式i-1化合物在不同钙离子浓度的1×pbs溶液中的荧光发射谱图;(b)为在不同钙离子浓度的1×pbs溶液中式i-1化合物在547nm处的荧光发射强度变化曲线;λex=351nm;
图6为式i-1化合物与钙离子结合后的高分辨质谱图,图中化学式为式i-1化合物与钙离子结合物的化学式;
图7为式i-1化合物在添加和未添加钙离子的不同金属离子(zn2+,li+,na+,k+,mg2+,cu2+,fe3+,co2+,ni2+)溶液中荧光强度的柱状图;图中“-”表示未添加钙离子;“+”表示添加钙离子;
图8为小鼠骨髓间充质干细胞在不同浓度的实施例1制备的式i-1化合物下的细胞存活率柱状图;
图9为实施例1制备的式i-1化合物染色的骨裂缝在不同染色时间下的荧光成像照片;
图10为实施例1制备的式i-1化合物染色的羟基磷灰石支架裂缝在不同染色时间下的荧光成像照片;
图11为钙黄绿素和实施例1制备的式i-1化合物分别染色的骨裂缝在洗涤前后的荧光照片;a~c对应钙黄绿素,d~f对应式i-1化合物;
图12为实施例1制备的式i-1化合物染色的脑膜瘤的钙化区的荧光照片。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
(1)式iv-1化合物的合成,其合成方程式如下::
5-苄氯水杨醛(340mg,2mmol),亚氨基二乙酸二乙酯(378mg,2mmol),n,n-二异丙基乙基胺(i-pr2etn)(330μl,258mg,2.0mmol)在乙腈(10ml)中回流反应2小时。待反应结束后,冷却至室温,减压蒸除溶剂,残余物通过硅胶柱层析分离(石油醚:乙酸乙酯=5:1)得到式iv-1化合物(288mg,89%)。1hnmr(cdcl3,500mhz)δ10.79(s,1h),9.72(s,1h),7.48(d,j=2.0hz,1h),7.43(dd,j1=8.5hz,j2=2.0hz,1h),6.77(d,j=8.5hz,1h),4.01(q,j=7.5hz,4h),3.74(s,2h),3.39(s,4h),1.11(t,j=7.5hz,6h);13cnmr(cdcl3,125mhz)δ196.5,170.9,160.9,137.8,133.9,129.9,120.3,117.5,60.4,56.5,54.0,14.1.
(2)式iii-1化合物的合成,其合成方程式如下:
将式iv-1化合物(332mg,1.0mmol)溶于无水乙醇(10ml)中,随后加入水合肼(25mg,0.5mmol)和50μl醋酸,在回流下反应15分钟;冷却至室温,过滤生成的固体,随后用乙醇洗涤两次(5ml×2),真空干燥后得到式iii-1化合物(235mg,81%)。1hnmr(cdcl3,500mhz)δ11.34(s,2h),8.70(s,2h),7.49–7.36(m,4h),7.00(d,j=8.5hz,2h),4.18(q,j=7.0hz,8h),3.88(s,4h),3.55(s,8h),1.28(t,j=7.0hz,12h);13cnmr(cdcl3,125mhz)δ171.0,164.7,159.3,134.5,133.1,129.4,117.2,117.1,60.6,56.9,54.1,14.3;hrms(maldi-tof):m/z[m+h]+calcdforc32h43n4o10:643.2979;found:643.2973.
(3)式ii-1化合物的合成,其合成方程式如下:
将式iii-1化合物(128mg,0.2mmol)溶于thf(5ml)中,随后加入氢氧化钠水溶液(5ml,0.8m).在室温(rt表示室温)下反应1小时后,加入1wt%稀盐酸,析出黄色固体,过滤真空干燥得到式ii-1化合物(57.9mg,55%)。1hnmr(500mhz,dmso-d6)δ12.26(s,4h),11.05(s,2h),8.98(s,2h),7.63(d,j=2.0hz,2h),7.39(dd,j1=8.5hz,j2=2.0hz,2h),6.95(d,j=8.5hz,2h),3.78(s,4h),3.42(s,8h);13cnmr(dmso-d6,125mhz)δ172.4,162.5,157.9,134.0,130.7,129.8,118.0,116.6,56.4,53.6;hrms(esi):m/z[m+na]+calcdforc24h26n4nao10:553.1547;found:553.1538。
(4)聚集诱导发光探针(式i-1化合物)的合成,其合成方程式如下:
将式ii-1化合物(26.5mg,0.05mmol)和甲醇钠(10.8mg,0.2mmol)溶于5ml甲醇中,在室温(rt表示室温)下搅拌0.5h;待反应结束后,减压蒸除反应溶剂,随后真空干燥得到式i-1化合物(30.3mg,98%)。1hnmr(500mhz,d2o)δ8.71(s,2h),8.46(s,2h),7.54(d,j=1.5hz,2h),7.38(dd,j1=8.5,j2=2.0hz,2h),6.92(d,j=8.5hz,2h),4.22(s,4h),3.70(s,8h);13cnmr(d2o,125mhz)δ170.1,164.5,159.2,135.7,135.3,120.7,117.9,117.4,57.8,55.9。
性能测试:
(一)将实施例1制备的式i-1化合物、式ii-1化合物以及式iii-1化合物进行光物理性质测试:
(1-1)式iii-1化合物的光物理性质表征:
将四氢呋喃和水按照不同的体积比(四氢呋喃:水=100:0,90:10,80:20,70:30,60:40,50:50,40:60,30:70,20:80,10:90,1:99)混合,形成含水量不同的混合液,将式iii-1化合物溶解到这些混合液中,使化合物的浓度为10-5mol·l-1,随后检测荧光发射光谱,结果见图1。图1为实施例1制备的式iii-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式iii-1化合物在四氢呋喃溶液中的归一化紫外吸收光谱图;(b)为式iii-1化合物在四氢呋喃和水的混合溶液中不断增加水含量的荧光发射光谱图;(c)为式iii-1化合物在四氢呋喃和水的混合溶液中不断增加水含量的最大荧光发射强度与四氢呋喃中最大荧光发射强度的比值变化图;λex=366nm。
从图中可知,在四氢呋喃和水的混合溶剂体系中增加水的比例至(四氢呋喃:水=10:80)时,式iii-1化合物的溶解度迅速降低,从而生成聚集体,由于分子内运动受限,荧光发射迅速增强。
(2-1)式ii-1化合物的光物理性质表征:
将二甲基亚砜和乙醇按照不同的体积比(二甲基亚砜:乙醇=100:0,90:10,80:20,70:30,60:40,50:50,40:60,30:70,20:80,10:90,1:99)混合,形成含乙醇量不同的混合液,将式ii-1化合物溶解到这些混合液中,使化合物的浓度为10-5mol·l-1,随后检测荧光发射光谱,结果见图2。图2为实施例1制备的式ii-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式ii-1化合物在二甲基亚砜溶液中的归一化紫外吸收光谱图;(b)为式ii-1化合物在二甲基亚砜和乙醇的混合溶液中不断增加乙醇含量的荧光发射光谱图;(c)为式ii-1化合物在二甲基亚砜和乙醇的混合溶液中不断增加乙醇含量的最大荧光发射强度与二甲基亚砜溶液中最大荧光发射强度的比值变化图;λex=363nm。
从图中可知,随着乙醇含量的增加,式ii-1化合物的溶解度逐渐降低,荧光发射逐渐增强。
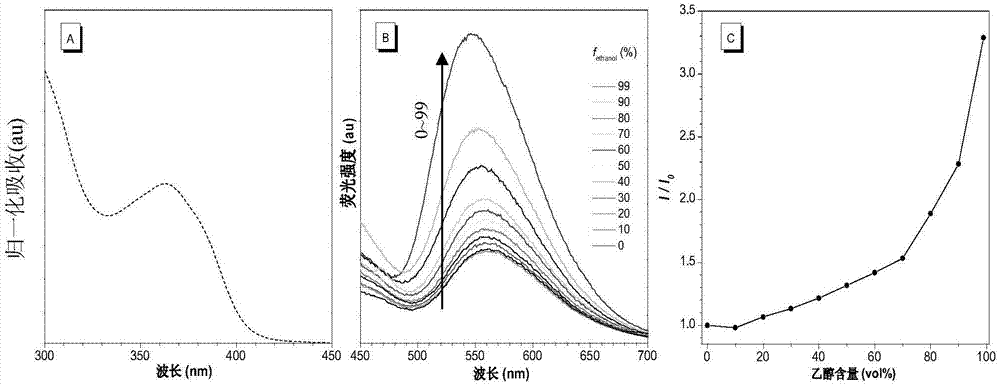
(3-1)式i-1化合物的光物理性质表征:
将水和四氢呋喃按照不同的体积比(水:四氢呋喃=100:0,90:10,80:20,70:30,60:40,50:50,40:60,30:70,20:80,10:90,1:99)混合,形成含四氢呋喃量不同的混合液,将式i-1化合物溶解到这些混合液中,使化合物的浓度为10-5mol·l-1,随后检测荧光发射光谱,结果见图3。图3为实施例1制备的式i-1化合物的紫外吸收光谱图和荧光发射光谱图以及荧光发射强度比值变化图;(a)为式i-1化合物在水溶液中的归一化紫外吸收光谱图;(b)为式i-1化合物在水和四氢呋喃的混合溶液中不断增加四氢呋喃含量的荧光发射光谱图;(c)为式i-1化合物在四氢呋喃和水的混合溶液中不断增加四氢呋喃含量的最大荧光发射强度与水溶液中最大荧光发射强度的比值变化图;λex=351nm。
从图中可知,在水和四氢呋喃的混合溶剂体系中增加四氢呋喃的比例至(四氢呋喃:水=10:90)时,式i-1化合物的溶解度迅速降低,从而生成聚集体,由于分子内运动受限,荧光发射迅速增强。
将通过测量式i-1化合物在水溶液中和薄膜态的量子产率和荧光寿命,发现相对于在水溶液态,式i-1化合物在薄膜态的量子产率增加了46.1倍(从0.23%增加至10.6%),荧光寿命增加了1.89倍(从1.32ns增加至2.48ns),清楚证实了其聚集诱导发光性质。
(二)考察ph对实施例1制备的式i-1化合物的紫外-可见吸收的影响
将实施例制备的式i-1化合物溶解至不同ph(ph=4.0,6.0,7.0,7.4)的水溶液中(式i-1化合物浓度10μm),随后测定其紫外-可见吸收光谱,测试结果如图4所示。图4为实施例1制备的式i-1化合物在不同ph的水溶液(ph=4.0,6.0,7.0,7.4)的归一化紫外吸收图。
通过光谱分析发现(图4),在不同ph环境下,式i-1化合物的紫外-可见吸收光谱未发生明显变化,表明化合物i-1在ph=4.0-7.4范围内具有较好的稳定性。
(三)考察钙离子对实施例1制备的式i-1化合物性能的影响
(3-1)钙离子浓度对实施例1制备的式i-1化合物的荧光性能的影响
将实施例1制备的式i-1化合物的1×pbs溶液(30μl,100mm)加入到2.97ml不同浓度的钙离子的1×pbs溶液(浓度为0,0.2,0.4,0.6,0.8,1.0,1.2,1.4,1.6,1.8,2.0,2.2,2.4,2.6,2.8,3.0mm)中,漩涡震荡1分钟,随后测定荧光光谱。图5为钙离子浓度对实施例1制备的式i-1化合物荧光性能的影响图;(a)为式i-1化合物在不同钙离子浓度的1×pbs溶液中的荧光发射谱图;(b)为在不同钙离子浓度的1×pbs溶液中式i-1化合物在547nm处的荧光发射强度变化曲线;λex=351nm。
从图中可知,式i-1化合物对钙离子检测具有宽响应范围和高点亮比率的检测能力。
(3-2)考察实施例1制备的式i-1化合物与钙离子的螯合情况
将实施例1制备的式i-1化合物的水溶液(1.0mm)与cacl2水溶液(2.0mm)混合,随后通过高分辨质谱鉴定其结合状态,测试结果如图6所示。从图中可知,实测值为607.0661,与理论值607.0666相符,表明式i-1化合物对钙离子有很好的鳌合能力。图6为实施例1制备的式i-1化合物与钙离子结合后的高分辨质谱图,图中化学式为式i-1化合物与钙离子结合物的化学式。
(3-3)考察实施例1制备的式i-1化合物对钙离子是否具有选择识别能力
将实施例制备的式i-1化合物分别与不同金属离子(zn2+,li+,na+,k+,mg2+,cu2+,fe3+,co2+,ni2+)混合,形成9个实验组;以未添加上述金属离子为对照组,测定实验组和对照组的荧光强度;向实验组和对照组中加入钙离子,再测定荧光强度,测试结果如图7所示。
图7为实施例1制备的式i-1化合物在添加和未添加钙离子的不同金属离子(zn2+,li+,na+,k+,mg2+,cu2+,fe3+,co2+,ni2+)溶液中荧光强度的柱状图。从图中可知,式i-1化合物对钙离子具有选择性识别能力。
(四)细胞毒性的检测
将不同浓度的实施例1制备的式i-1化合物(0,25,50,100,200,300,400和500μm)与小鼠骨髓间充质干细胞(mbmscs)混合,测定细胞的存活率,测试结果如图8所示。图8为小鼠骨髓间充质干细胞在不同浓度的实施例1制备的式i-1化合物下的细胞存活率柱状图。从图中可知,式i-1化合物具有很好的生物相容性,在不同浓度下(0,25,50,100,200,300,400和500μm)都几乎没有细胞毒性,适于生理环境下对钙离子的检测。
(五)实施例1制备的式i-1化合物的特异性成像
(5-1)骨裂缝特异性成像
在实施例1制备的式i-1化合物(500μm)水溶液中染色骨裂缝,观察式i-1化合物对骨裂缝的特异性成像情况(即不同染色时间下,骨裂缝的荧光成像情况),观察结果如图9所示。图9为实施例1制备的式i-1化合物染色的骨裂缝在不同染色时间下的荧光成像照片。λex=405nm,λem=460-750nm。从图中可知,式i-1化合物对骨裂缝具有特异性成像能力。在染色时间为0时(即骨裂缝未染色),骨裂缝无荧光成像;染色时间为0.5h、1h、2h、3h、4h,骨裂缝都能够明显地荧光成像。
(5-2)羟基磷灰石支架裂缝特异性成像
在实施例1制备的式i-1化合物(500μm)水溶液中染色羟基磷灰石支架上的裂缝,观察式i-1化合物对羟基磷灰石支架上的裂缝的特异性成像情况(即不同染色时间下,羟基磷灰石支架上的裂缝的荧光成像情况),观察结果如图10所示。图10为实施例1制备的式i-1化合物染色的羟基磷灰石支架裂缝在不同染色时间下的荧光成像照片。λex=405nm,λem=460-750nm。从图中可知,式i-1化合物对羟基磷灰石支架上的裂缝具有特异性成像能力。在染色时间为0时(即羟基磷灰石支架上的裂缝未染色),羟基磷灰石支架上的裂缝无荧光成像;染色时间为0.5h、1h、2h、3h、4h,羟基磷灰石支架上的裂缝都能够明显地荧光成像。
(5-3)背景干扰
在钙黄绿素水溶液(500μm)中和实施例1制备的式i-1化合物(500μm)水溶液中分别染色骨裂缝,染色一段时间后(染色1h),对骨裂缝进行洗涤,观察洗涤前后骨裂缝的荧光成像,测试结果如图11所示。图11为钙黄绿素和实施例1制备的式i-1化合物分别染色的骨裂缝在洗涤前后的荧光照片。钙黄绿素染色的骨裂缝洗涤前的荧光图为图11(b),经过多次洗涤荧光图为图11(c);式i-1化合物染色的骨裂缝洗涤前后荧光图一致(图11(e)、图11(f))。图11(a)为钙黄绿素处理的明场图,图11(d)为式i-1化合物处理的明场图。实验结果可知,钙黄绿素具有高的背景噪音,需要多次洗涤才能除去背景干扰;而式i-1化合物染色的骨裂缝洗涤前后作为免洗荧光探针,不需要洗涤,可直接用于骨裂缝的特异性荧光成像。
(六)钙化斑块的特异性成像
采用实施例1制备的式i-1化合物对脑膜瘤的钙化区进行染色,染色步骤为:(1)脱蜡:二甲苯中脱蜡5min,再换新鲜的二甲苯,脱蜡5min,最后再换并脱蜡5min;(2)梯度入水:95%,70%,30%的乙醇中各2min,超纯水2min;(3)将式i-1化合物用超纯水配制成10mm溶液,并且以样品覆盖切片样本,孵育3.5h,然后用超纯水缓慢冲洗玻片;(4)通过共聚焦荧光显微镜对脑膜瘤的钙化区进行荧光拍照。测试结果如图12所示。图12为实施例1制备的式i-1化合物染色的脑膜瘤的钙化区的荧光照片。该实验结果表明,式i-1化合物作为荧光探针可特异性染色脑膜瘤的钙化区。
- 还没有人留言评论。精彩留言会获得点赞!