一种乙螨唑制备方法与流程
本发明属于农药杀虫剂领域,涉及一种乙螨唑制备方法。
背景技术:
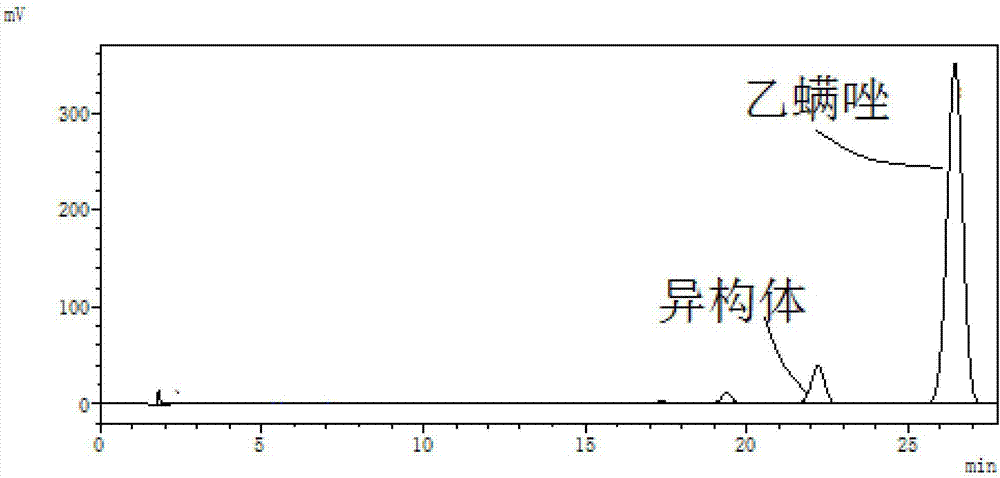
:乙螨唑,英文名:etoxazole,为1994年日本八洲化学公司研发的一种高效低毒农用杀螨剂,具有二苯基噁唑啉结构的新型化合物,其化学名为4-(4-(叔丁基)-2-乙氧基苯基)-2-(2,6-二氟苯基)噁唑啉,分子式为:c21h23f2no2,cas号为:153233-91-1;外观白色晶体粉末,熔点:101-102℃,结构式如下:目前,乙螨唑的制备方法均是通过中间体分子内环合而来,cn1024794、us5478855和ep0639572分别公开了两种制备乙螨唑的方法:路线1中的脱水剂通常为硫酸、氯化亚砜、三氯氧磷、五氧化二磷,其反应条件要求比较苛刻,产生的三废较多,不适合工业化生产。路线2采用甲醇、乙醇、n,n'-二甲基甲酰胺或二甲基亚砜作溶剂,氢氧化钠水溶液、氢氧化钾或碳酸钾做碱进行环合反应,条件温和,副产物仅为无机盐,适合工业化生产,然而该方法还存在两点不足,第一点:由于酰胺中的o原子和n原子均具有亲核性,o原子参与环合则生成目标产物乙螨唑,n原子参与环合则生成杂质异构体,us5478855和ep0639572公开了氯化、环合两步反应的收率仅62.5%。另外,目前有公开的乙螨唑合成(新颖杀螨剂-乙螨唑的合成,戴炜锷、程志明,浙江化工,2009年第40卷第7期7-9页)报道中,利用20%氢氧化钠溶液做碱制备乙螨唑,环合步骤收率为70%。第二点:采用氢氧化钠水溶液做碱,则溶剂回收中需要经过精馏才能将甲醇和水分离,生产的设备成本与能耗将大大增加。因此,为了克服现有方法的缺陷,需要开发新的乙螨唑制备方法。技术实现要素:针对现有技术的不足,本发明的目的在于提供一种乙螨唑制备方法。为达到此发明目的,本发明采用以下技术方案:本发明提供一种乙螨唑制备方法,所述制备方法为式i所示化合物与氨气或氨溶液反应生成乙螨唑,具体反应式如下所示:此类反应常规手段是利用强碱进行环合反应,而本方法突破常规手段,采用弱碱进行反应,能够降低异构体的生成,选择性好、收率高、产品含量高,副产物仅有氯化铵,并且达到工业用氯化铵的国标要求,适于工业化生产。优选地,相对于1当量的所述式i所示化合物,所述氨气或氨溶液的当量为1~10,例如可以是1当量、2当量、3当量、4当量、5当量、6当量、8当量、9当量或10当量。优选地,所述氨溶液为氨甲醇溶液或氨水。优选地,所述反应的溶剂为甲醇、乙醇、n,n'-二甲基甲酰胺或二甲基亚砜中的一种或至少两种的组合,进一步优选甲醇和/或乙醇。优选地,所述反应的温度为70~120℃,例如可以是70℃、80℃、90℃、100℃、110℃或120℃。优选地,所述反应的时间为5~20小时,例如可以是5小时、8小时、10小时、12小时、15小时、18小时或20小时。优选地,所述反应在高压釜中进行。优选地,所述反应的压力为0.1~0.5mpa,例如可以是0.1mpa、0.2mpa、0.3mpa、0.4mpa或0.5mpa。作为本发明的优选技术方案,乙螨唑的制备方法为:将式i所示的化合物加入到高压釜中,用溶剂溶解,相对于1当量式i所示的化合物,加入1~10当量的氨气或氨溶液,在70~120℃、0.1~0.5mpa下反应5~20小时,得到所述乙螨唑。相对于现有技术,本发明具有以下有益效果:本发明利用弱碱氨气或氨溶液反应,突破了以氢氧化钠等强碱反应的常规手段,达到了非常良好的效果。反应选择性好,可大幅降低异构体杂质的生成,反应收率高达87-95%,产品含量高达95-97%,副产物仅为氯化铵,含量可达99%以上,符合工业用氯化铵的国标要求;并且若采用氨气和氨甲醇溶液又能避免将水引入反应体系,从而省去甲醇与水的精馏分离过程,降低了生产设备的成本与能耗,适合于工业化生产。附图说明图1是本发明实施例1中的乙螨唑hplc谱图。图2是本发明对照例1中的乙螨唑hplc谱图。具体实施方式下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。本发明中乙螨唑含量检测方法采用高效液相色谱外标法,乙螨唑标准品来源:dr.ehrenstorfergmbh。氯化铵含量检测方法:参考gb/t2946-2008。实施例1在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,100g无水甲醇,关闭釜盖,打开进料阀通入氨气4.2g后,关闭阀门升温至90℃,釜压0.2mpa,搅拌反应8小时后,撤去热源,取样,高效液相色谱中控(条件:250mm×4.6mm(i.d)不锈钢柱,内装c18(5μm)填充物;流动相:乙腈:水=70:30(体积比);流速:1.0ml/min;波长:225nm),中控谱图如图1所示,具体数据如表1所示,通过液相色谱质谱联用技术,分别得到保留时间20.8min、保留时间24.3min的化合物分子量(高分辨质谱),数据如表1所示。反应完全后,开启泄压阀,连接真空泵,体系压力5-10kpa,回收50g甲醇后,撤去真空,降至0-5℃结晶1小时,在此过程中有大量白色固体析出,过滤、滤饼烘干后用90g甲苯与30g水溶解,分层后,水层脱干得6.3g白色固体,即为副产氯化铵,含量99.5%,氯化铵的重量收率96.1%;有机层蒸除甲苯后得40.5g乙螨唑白色固体,含量为97%,乙螨唑重量收率92%。表1峰号保留时间化合物名hrms[m+h]+归一面积/%120.8min异构体360.17531.0224.3min乙螨唑360.175496.7实施例2在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,150g无水乙醇,关闭釜盖,打开进料阀通入氨气6.2g后,关闭阀门升温至80℃,釜压0.1mpa搅拌反应7小时后,撤去热源,取样,高效液相色谱中控,异构体的归一面积1.2%,乙螨唑归一面积96%。待反应液降至60℃后,趁热过滤,滤饼烘干得6g白色固体,即为副产氯化铵,含量99.3%,氯化铵的重量收率91.6%。滤液减压蒸馏,体系压力5-10kpa,回收100g乙醇后,撤去真空,釜内浓缩液降温至0-5℃结晶1小时,在此过程中有大量白色固体析出,过滤、滤饼烘干得40.1g乙螨唑白色固体,含量为96%,乙螨唑重量收率91%。实施例3在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,120gn,n'-二甲基甲酰胺,关闭釜盖,打开进料阀通入氨气2.1g后,关闭阀门升温至70℃,氮气补压至0.4mpa,搅拌反应20小时后,撤去热源,取样,高效液相色谱中控,异构体的归一面积0.7%,乙螨唑归一面积93%。开启泄压阀,连接真空泵,体系压力1-3kpa,减压蒸馏回收溶剂,釜内残留物加入90g甲苯与30g水溶解,分层后,水层脱干得5.9g白色固体,即为副产氯化铵,含量99.1%,氯化铵的重量收率90%;有机层蒸除45g甲苯后,降温至0-5℃结晶得38.3g固体,含量为95%,乙螨唑重量收率87%。实施例4在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,200g二甲基亚砜,关闭釜盖,打开进料阀通入氨气10.4g后,关闭阀门升温至120℃,氮气补压至0.3mpa,搅拌反应5小时后,撤去热源,取样,高效液相色谱中控,异构体的归一面积1.1%,乙螨唑归一面积96%。开启泄压阀,连接真空泵,体系压力1-3kpa,减压蒸馏回收溶剂,釜内残留物加入90g甲苯与30g水溶解,分层后,水层脱干得6.1g白色固体,即为副产氯化铵,含量99.5%,氯化铵的重量收率93%;有机层蒸除45g甲苯后,降温至0-5℃结晶得41g固体,含量为96%,乙螨唑重量收率93%。实施例5(1)氨甲醇溶液的制备在500ml三口瓶中,加入200g无水甲醇,放入0℃浴槽中,待降至5℃,开始液下通入氨气。2小时后,即得氨的甲醇溶液,通过增重法计算氨气在甲醇溶液的浓度为16%(重量百分比)。(2)乙螨唑的制备在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,130g上述氨甲醇溶液,关闭阀门升温至100℃,釜压0.2mpa,搅拌反应13小时后,撤去热源,取样,高效液相色谱中控,异构体的归一面积1.0%,乙螨唑归一面积97%。开启泄压阀,连接真空泵,体系压力5-10kpa,回收60g甲醇后,撤去真空,继续降至0-5℃结晶1小时,在此过程中有大量白色固体析出,过滤、滤饼烘干后用90g甲苯与30g水溶解,分层后,水层脱干得6.2g白色固体,即为副产氯化铵,含量99.6%,氯化铵的重量收率95%;有机层蒸除甲苯后得41.4g乙螨唑白色固体,含量为97%,乙螨唑重量收率94%。实施例6在500ml不锈钢高压釜中加入50g(含量97%)原料式i所示化合物,150g甲醇,66g氨水(含量25%),关闭阀门升温至110℃,釜压0.5mpa,搅拌反应16小时后,撤去热源,取样,高效液相色谱中控,异构体的归一面积1.1%,乙螨唑归一面积96%。继续降温至5-10℃结晶1小时,在此过程中有大量白色固体析出,过滤、滤饼用水淋洗两次后,烘干得41.8g乙螨唑白色固体,含量为95%,乙螨唑重量收率95%。滤液常压蒸馏,待气相温度达到100℃后,停止蒸馏,加入10g甲苯萃取回收少量的乙螨唑,剩余水层脱干得6g白色固体,即为副产氯化铵,含量99.1%,氯化铵的重量收率91.6%对照例1在500ml三口瓶中加入40.2g(含量97%)原料式i所示化合物,158g无水甲醇,然后缓慢滴入85g质量浓度为20%的氢氧化钠水溶液,滴毕后混合物升温至70℃,搅拌反应1小时,hplc中控(条件:250mm×4.6mm(i.d)不锈钢柱,内装c18(5μm)填充物;流动相:乙腈:水=70:30(体积比);流速:1.0ml/min;波长:225nm),高效液相色谱谱图见图2,具体数据如表2所示,通过液相色谱质谱联用技术,分别得到保留时间22.2min、保留时间26.4min的化合物分子量(高分辨质谱),数据如表2所示。减压蒸馏回收甲醇和水混合溶液,釜内残留物加入75g甲苯与30g水溶解,分层后,有机层蒸除40g甲苯后,降温至0-5℃结晶得27.3g固体,含量为95%,乙螨唑重量收率77%。表2对比实施例1和对照例1,可以得出的结论是,采用本发明的制备方法制备得到的乙螨唑收率和含量均较高,抑制异构体杂质效果非常明显,而采用naoh水溶液法制备得到的乙螨唑收率低,并且异构体杂质较多。因此本发明的制备方法具有收率高、含量高的优点,适于工业化生产。申请人声明,本发明通过上述实施例来说明本发明的一种乙螨唑制备方法,但本发明并不局限于上述工艺步骤,即不意味着本发明必须依赖上述工艺步骤才能实施。所属
技术领域:
的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1