一种提高谷胱甘肽累积量的生物方法与流程
本发明属于生物工程技术领域,具体地说,是关于一株谷胱甘肽合成酶系与表达胱氨酸转运系统底物结合蛋白相构建的基因缺陷型菌株高产谷胱甘肽的方法。
背景技术:
谷胱甘肽(γ-l-glutamyl-cysteinyl-glycine,gsh),是由l-谷氨酸、l-半胱氨酸和甘氨酸组成的生物活性三肽化合物,是细胞内重要的抗氧化剂和主要的非蛋白巯基化合物(>90%)。在生物组织中,谷胱甘肽通过谷胱甘肽还原酶保持氧化型和还原型的动态平衡并发挥细胞保护、物质代谢、信息调控、调节细胞及组织的发育、对抗或诱发疾病等作用,因此,大量应用于医药保健、护肤美容、食品添加等行业。
目前gsh的生产方法主要有溶剂萃取法、化学合成法和生物酶催化法和生物发酵法。相比之下,生物发酵法具有反应条件温和、反应步骤简单、成本低、转化效率高、生产速率快等优势,是今后生产谷胱甘肽的主要趋势,但目前大部分研究还停留在实验室阶段,实现商业化生产的国家主要是日本。
近来分子生物学的发展也为研究者从分子水平探究、提高gsh产量提供了新思路。l-半胱氨酸是合成谷胱甘肽的前体氨基酸之一,细胞内l-半胱氨酸的供应不足严重影响了谷胱甘肽的合成速度及产量。因此可以采用分子生物学方法使l-半胱氨酸大量进入细胞,最终使细胞内谷胱甘肽的合成量增加。此外,同时利用生物体的代谢反应,抑制降解途径可有效提高谷胱甘肽产量。
技术实现要素:
本发明的目的在于,提供一种谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒,以用于构建表达谷胱甘肽合成酶系与表达胱氨酸转运系统底物结合蛋白的基因缺陷型菌株。
本发明还有一个目的在于,提供一种谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒的构建方法。
本发明还有一个目的在于,提供一种谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒的应用。
本发明还有一个目的在于,提供一种抑制谷胱甘肽降解途径的方法。
本发明提供的谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒包括gsh1,gsh2和fliy序列和两个合适的载体片段;所述gsh1,gsh2和fliy序列分别如ncbi上geneid为242378250,253978924和948833的序列所示。
根据本发明的一个优选实例,在谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的共表达重组质粒中,gsh和fliy编码区置于t7启动子和lac操纵子之间。
根据本发明的另一个优选实施例,所述谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒分别包括一个卡那霉素抗性基因和氨苄青霉素抗性基因,用以筛选基因重组菌。
本发明提供的谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒的构建方法包括以下步骤:
a)pcr扩增获得gsh1,gsh2和fliy序列;
b)将gsh1和fliy序列共同克隆入prsfduet-1,gsh2克隆入pbad24。
本发明提供的谷胱甘肽合成酶系与胱氨酸转运系统底物结合蛋白构成的重组质粒可用于构建表达谷胱甘肽合成酶系与表达胱氨酸转运系统底物结合蛋白的基因缺陷型菌株。
本发明提供的方法可用于累积谷胱甘肽。
本发明提供的累积谷胱甘肽的方法通过发酵培养本发明提供的谷胱甘肽合成酶系与表达胱氨酸转运系统底物结合蛋白的基因缺陷型菌株,累积谷胱甘肽。
根据本发明的另一个优选实施例,累积谷胱甘肽的方法还通过,在选择高产菌株后,通过高密度发酵的技术来累积谷胱甘肽。
使用本发明提供的谷胱甘肽合成酶系与表达胱氨酸转运系统底物结合蛋白的基因缺陷型菌株,可以明显地提高谷胱甘肽生产量,说明此方法,提高了原料利用率,降低了生产成本和能耗,为进一步研究谷胱甘肽产量建立一种方法,为其产业化生产奠定一种基础。
附图说明
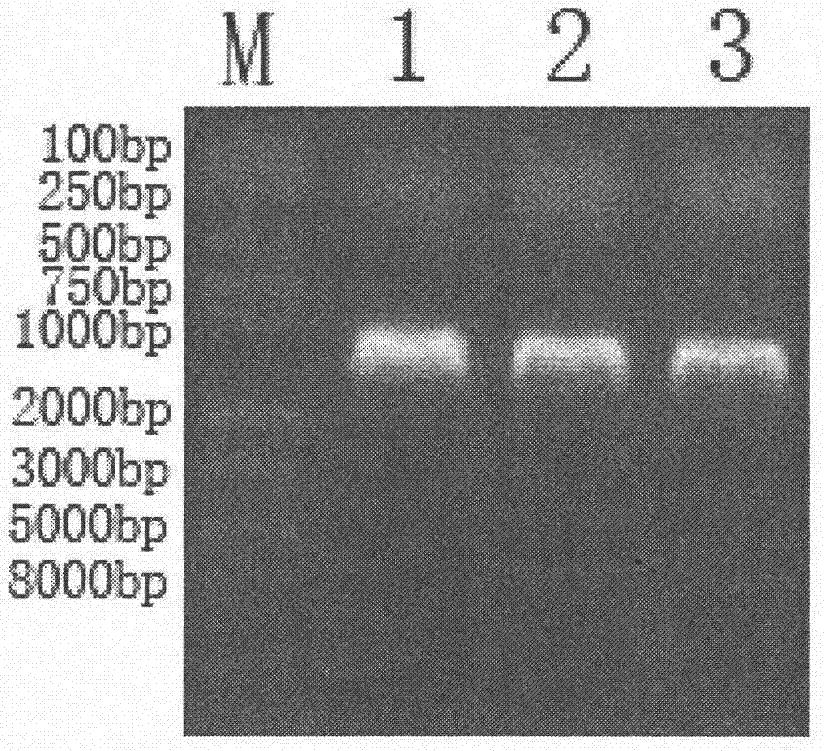
图1是pcr扩增获得的gsh1,gsh2和fliy的检测结果,其中泳道1为gsh1,泳道2为gsh2,泳道3为fliy。
图2是pcr扩增获得的pkd3-cm的检测结果,其中泳道1为pkd3-cm。
图3是抗性基因cm去除的pcr鉴定结果,其中泳道1为pept的pcr扩增产物,泳道2为apept的pcr扩增产物。
具体实施方式
以下结合具体实施例,对本发明作进一步说明。应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。
以下实施例中为注明具体条件的实验方法,通常按照常规条件,如《分子克隆:实验室手册》(newyork:coldspringharborlaboratorypress,1989)中所述的条件或厂商提供的方案进行。
在本发明的下述实施例中,使用的胶回收试剂盒、质粒提取试剂盒、基因组提取试剂盒均购自生工生物公司。
在本发明的下述实施例中,使用的pmd-19tsimplevector,bamhi,xbai,hindiii,ndei,ecorv,taqdna聚合酶,t4dna连接酶,dnamarker,均购自takara公司。
在本发明的下述实施例中,使用的表达载体prsfduet-1,pbad24,菌种e.colibl21(de3),e.colidh5α,为中国药科大学生命中心实验室保存,其中菌种e.colibl21(de3)的atcc编号为baa-1025tm。
在本发明的下述实施例中,使用的e.colibl21(de3)感受态细胞、e.colidh5α感受态细胞,按照invitrogen公司pichiaexpressionkit手册中提供的方法进行。
在本发明的下述实施例中,使用的lb培养基的配方为:1%胰蛋白胨,0.5%酵母浸粉,1%nacl;使用的摇瓶发酵培养基的配方为:1%胰蛋白胨,0.5%酵母浸粉,1%nacl。配置固体lb平板培养基时,按照上述配方添加2%琼脂粉。
在本发明的下述实施例中,感受态细胞的制备和转化,按照《分子克隆:实验室手册》提供的方法进行。
实施例1两个表达质粒的构建
1.1引物设计
根据ncbi报道的gsh1,gsh2和fliy序列,设计以下6条引物:
gsh1up:cgcggatccatgatcccggacgtatcacaggc;
gsh1down:cccaagctttcaggcgtgtttttccagccacacc;
gsh2up:gctctagagatgatcaagctcggcatcgt;
gsh2down:atgaagcttttactgctgctgtaaacgtgc;
fliyup:gggcatatgatgaaattagcacatctg;
fliydown:cccgatatcttatttggtcacatcagc。
其中gsh1up和gsh1down用于扩增gsh1编码区;gsh2up和gsh2down用于扩增gsh编码区;fliyup和fliydown用于扩增fliy编码区;gsh1up、gsh1down、gsh2up、gsh2down、fliyup和fliydown上的下划线表示在gsh1up、gsh1down、gsh2up、gsh2down、fliyup和fliydown上分别引入的bamhi、hindiii、xbai、hindiii、ndei和ecorv酶切位点。
1.2pcr扩增gsh1,gsh2和fliy序列
抽提得到大肠杆菌e.colik12基因组。
以大肠杆菌e.colik12基因组为模板,分别以步骤1.1中设计的gsh1up和gsh1down,gsh2up和gsh2down以及fliyup和fliydown为引物对,进行pcr扩增,具体如下:
扩增gsh1,gsh2和fliy的反应体系均为:premixtaqdna聚合酶25μl,e.colik12基因组dna1μl,上下游引物(10μm)各1μl,加入去离子水20μl,至体系总体积为50μl。
扩增反应条件均为:94℃4min;94℃30s,55℃30s,72℃1min,34个循环;72℃10min;4°∞,30个循环
pcr扩增产物分别进行凝胶电泳检测,检测结果如图1所示。获得的产物的大小分别为1500bp,900bp和800bp,符合gsh1,gsh2和fliy产物的预期大小。
1.3表达载体的构建
1.3.1构建预处理
使用胶回收试剂盒纯化步骤1.2中获得的pcr产物,然后和pmd-19tsimple载体,用t4dna连接酶,于16℃连接过夜,反应体系如下:
4μl的pcr产物,1μlpmd-19tsimple载体,5μlt4dna连接酶。
连接产物转化大肠杆菌dh5α并涂布含有50μg/ml氨苄青霉素的固体lb平板,37℃培养至转化子长出。其中e.colidh5α是无氨苄青霉素抗性菌株,不能在含有氨苄青霉素的固体lb平板上生长,因此,平板上长出的转化子均为转化了pmd-gsh1,pmd-gsh2或pmd-fliy质粒的大肠杆菌。挑取转化子鉴定,根据鉴定结果,最终获得质粒pmd-gsh1,pmd-gsh2和pmd-fliy。
质粒pmd-gsh1,pmd-gsh2和pmd-fliy经测序验证,分别包含gsh1、gsh2和fliy编码区(gsh1、gsh2和fliy编码区序列分别如sqidno.1,sqidno.2和sqidno.3所示),而且编码区无氨基酸残基突变。
1.3.2表达载体的构建
将1.3.1中双酶切产物gsh1和gsh2凝胶回收纯化,分别与bamhi/hindiii双酶切的表达载体prsfduet-1和xbai/hindiii双酶切的表达载体pbad24连接,将连接产物分别转化入宿主菌e.colidh5α,并涂布含有60μg/ml卡那霉素和氨苄青霉素的固体lb平板,37℃培养至转化子长出。其中e.colidh5α是无卡那霉素或氨苄青霉素抗性菌株,不能在含有卡那霉素或氨苄青霉素的固体lb平板上生长,因此,平板上长出的转化子均为转化了prsfduet-1-gsh1或pbad24-gsh2质粒的大肠杆菌。挑取转化子鉴定,根据鉴定结果,最终获得两个质粒prsfduet-1-gsh1和pbad24-gsh2。通过ndei/ecorv酶切质粒pmd-fliy,将得到fliy表达框,然后将其连入使用ndei和ecorv酶切的质粒prsfduet-1-gsh1,获得质粒prsfduet-1-gsh1-fliy。
质粒prsfduet-1-gsh1-fliy和pbad24-gsh2经测序验证,分别包含gsh1,gsh2和fliy编码区(gsh1、gsh2和fliy编码区序列分别如sqidno.1,sqidno.2和sqidno.3所示),其中gsh1,gsh2和fliy编码区置于t7启动子、lac操纵子之下,而且编码区无氨基酸残基突变。
实施例2pept基因敲除菌株的构建
2.1引物设计
敲除pept基因共需4条引物,用于同源重组的引物s1、s2及用于鉴定缺陷株的引物s3、s4。
s1:
atggataaactacttgagcgatttttgaactacgtgtctctggatacccagtgtaggctggagctgc
s2:
ttacttccgttgcgccgttaactcggcaatacggacgatcacctgcaccgatgggaattagccatgg
s3:atggataaactacttgagcg
s4:ttacttccgttgcgccgtta
2.2基因缺陷株δpept的构建
同源重组辅助质粒pkd46转入宿主菌e.colibl21(de3),l-阿拉伯糖诱导后制备电转化感受态细胞。
以含氯霉素抗性基因cm的质粒pkd3为模板,s1、s2为引物进行pcr扩增反应,凝胶电泳检测结果如图2所示,获得的产物的大小为1230bp,与理论值一致,表明片段pkd3-cm扩增成功。
于冰上将2μl抗性片段pkd3-cm加入电转化感受态细胞,混匀后置于1mm电击杯中进行电击转化。电击参数为:2.5kv,脉冲3ms。电击完成后立即加入无抗性lb液体培养基,于37℃培养3h,使接入抗性片段的细胞充分复苏。然后取菌液涂布至含cm的lb平板,筛选出含抗性基因的缺陷株阳性克隆。将阳性单克隆分别接入含cm及amp的培养基中,42℃培养4-5h,筛选出在cm中正常生长,在amp中不能生长的菌种,即为重组成功的菌株δpept-cm。
将编码flp重组酶的质粒pcp20化学转化到菌株δpept-cm中,30℃培养后涂布氨苄抗性平板并筛选出转化子,然后转接到不含抗生素的lb液体培养基中42℃过夜培养。在消除cm的同时,对温度敏感的质粒pkd46、pcp20在高温下会逐渐丢失。消除质粒的菌株为基因缺陷株阳性克隆。
将阳性单克隆分别接入含cm及amp的培养基中,42℃培养4-5h,筛选出在cm和amp中都不能生长的菌种,该菌种即为完成敲除目的基因的菌种。为进一步查看抗性基因cm的去除情况,以阳性单克隆为模板,s3、s4为引物进行pcr扩增鉴定,凝胶电泳检测结果如图3所示,pcr产物长度为130bp,对照野生株pcr产物为1201bp,二者差异显著,证明基因缺陷株δpept构建成功。
实施例3表达质粒转化合成谷胱甘肽菌株
将质粒prsfduet-1-gsh1-fliy和pbad24-gsh2转化到基因缺陷株δpept,并涂布同时含有60μg/ml卡那霉素和氨苄青霉素的固体lb平板,37℃培养至转化子长出。其中基因缺陷株δpept是无卡那霉素和氨苄青霉素抗性菌株,不能在含有卡那霉素和氨苄青霉素的固体lb平板上生长,因此,平板上长出的转化子均为转化了prsfduet-1-gsh1-fliy和pbad24-gsh2质粒的大肠杆菌。
分别随机挑取若干含prsfduet-1-gsh1-fliy和pbad24-gsh2质粒的转化子,摇瓶发酵培养,并选取一株谷胱甘肽合成量较高的大肠杆菌菌株,命名为δpept-gsh12-fliy。
同时,将质粒prsfduet-1-gsh1-fliy和pbad24-gsh2,及prsfduet-1-gsh1和pbad24-gsh2两个组合分别转化到宿主菌e.colibl21(de3),并涂布同时含有60μg/ml卡那霉素和氨苄青霉素的固体lb平板,37℃培养至转化子长出。其中e.colibl21(de3)是无卡那霉素和氨苄青霉素抗性菌株,不能在含有卡那霉素和氨苄青霉素的固体lb平板上生长,因此,平板上长出的转化子均为分别转化了prsfduet-1-gsh1-fliy和pbad24-gsh2,及prsfduet-1-gsh1和pbad24-gsh2两个组合的大肠杆菌。
分别随机挑取若干含两个组合质粒的转化子,摇瓶发酵培养,并分别选取谷胱甘肽累积量较高的大肠杆菌菌株各两株,一株命名为bl21-gsh12-fliy,另一株命名为bl21-gsh12。
抽提δpept-gsh12-fliy,bl21-gsh12-fliy和bl21-gsh12的质粒dna,分别以引物gsh1up和gsh1down、gsh2up和gsh2down,fliyup和fliydown作为引物对,对上述抽提物进行pcr扩增,获得了长度为1500bp,900bp和800bp的dna片段,长度为1500bp和900bp的片段。经测序验证,gsh1的表达框插入了质粒prsfduet-1-gsh1,gsh1和fliy表达框已插入质粒prsfduet-1-gsh1-fliy,gsh2的表达框插入了质粒pbad24-gsh2。
根据上述结果,质粒prsfduet-1-gsh1-fliy,pbad24-gsh2和prsfduet-1-gsh1已成功转化入基因缺陷株δpept与宿主菌e.colibl21(de3),分别命名为δpept-gsh12-fliy,bl21-gsh12-fliy和bl21-gsh12。
实施例4δpept-gsh12-fliy,bl21-gsh12-fliy,bl21-gsh12和e.colibl21(de3)摇瓶发酵谷胱甘肽
将于37℃固体lb平板上生长过夜的δpept-gsh12-fliy,bl21-gsh12-fliy,bl21-gsh12和e.colibl21(de3)(原始菌,命名为bl21)的单菌落,分别接入液体lb培养基中,摇瓶培养过夜。
分别取适量菌液转接入摇瓶发酵培养基,培养10h。其中,在适当时间加入诱导剂;适时添加前体氨基酸并于发酵的3、5、7、9、11h取样,检测发酵产物中谷胱甘肽的累积量,检测结果如表1所示。
根据表1的结果,δpept-gsh12-fliy,bl21-gsh12-fliy,bl21-gsh12和e.colibl21(de3)中的谷胱甘肽的累积量,在发酵11h时达到最高且以δpept-gsh12-fliy效果最好。因此,在下一实施例中,仅选取摇瓶发酵后期累积量最高的δpept-gsh12-fliy进行发酵罐发酵。
表1δpept-gsh12-fliy,bl21-gsh12-fliy,bl21-gsh12和e.colibl21(de3)谷胱甘肽的累积量随培养时间的变化
实施例5δpept-gsh12-fliy发酵罐发酵谷胱甘肽
将于固体lb(含有60μg/ml卡那霉素)平板上长出的bl21-gsh12单菌落,接种至发酵罐发酵培养基,摇瓶培养过夜;取1ml接种至100ml发酵罐发酵培养基,摇瓶培养2h左右;然后接入1l的发酵罐发酵培养基中(2.5l发酵罐),并添加消泡剂聚氧丙稀聚氧乙烯甘油醚(即泡敌gpe),发酵罐发酵培养,发酵过程中适时添加适当浓度的诱导剂,并流加满足细胞生长需要的甘油,同时,通过流加氨水,控制ph为6.4;发酵过程中添加前体物质半胱氨酸,发酵30h停止发酵。
采用上述方法,进行平行实验,并分别取样检测发酵产物中的谷胱甘肽累积量,获得的重组菌发酵培养,发酵产物中原始菌株中谷胱甘肽的累积量,远远低于转化prsfduet-1-gsh1-fliy和pbad24-gsh2质粒的基因缺陷株δpept,但是通过将质粒prsfduet-1-gsh1-fliy和pbad24-gsh2转化累积谷胱甘肽的原始菌e.colibl21(de3),获得重组菌,发酵该重组菌,在适合的时间添加诱导剂iptg和阿拉伯糖,提高谷胱甘肽的方法,同样属于本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!