数字PCR方法快速检测羊水细胞非整倍体与流程
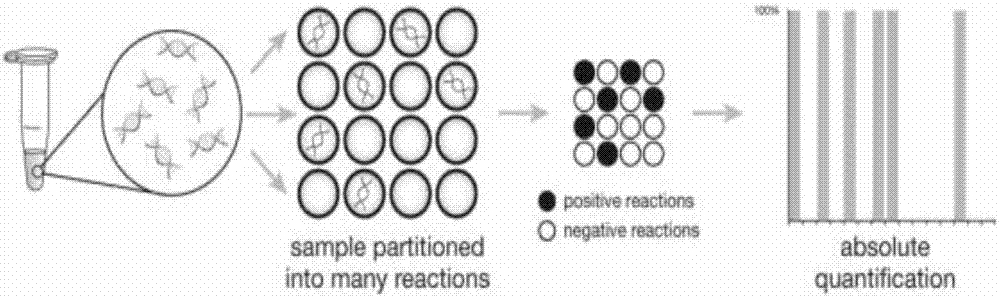
本发明涉及数字pcr方法快速检测羊水细胞非整倍体,具体涉及检测染色体非整倍体的扩增组合物,及快速检测试剂盒,以及检测方法,通过常染色体与目标染色体(13,18,21,x,y)之间计数的差异确定目标染色体的倍性情况,属于生物医疗
技术领域:
。
背景技术:
:引起新生儿缺陷率高的原因主要有以下几个方面:首先,与孕妇自身有关,随着孕妇年龄的增加,自身生理疾病或者染色体的异常变异几率增加,心理上的障碍因素都在一定程度上影响着胎儿的健康成长。其次,与生活环境也是密切相关的,雾霾、大气污染都会成为影响新生儿健康发育的因素。因此产前检测也成为控制人口缺陷的必要手段。染色体数目异常和结构畸变是导致出生缺陷的重要原因之一。染色体的数目异常和形态结构畸变,可以发于每一条染色体上。最常见的染色体疾病如唐氏综合征(21-三体),在新生儿中21-三体综合征的发病率约为1/800,但男性患儿多于女性,母亲年龄是影响发病率的重要因素。先天愚型患儿出生时体重和身长偏低,肌张力低下,突出的是颅面部畸形,头颅小而圆,脸圆而扁平,鼻扁平,眼距过宽,约1/2以上的患者有先天性心脏病。爱德华式综合征(18-三体)会导致严重畸形,在出生后不久死亡,发病率约为1:3500~8000新生儿,但在某些地区或季节明显增高,达到1:450~800,患儿中女性:男性比为4:1。患儿出生时体重低,吸吮差,反应弱,头面部和手足有严重畸形,头长而枕部凸出,面圆,眼距宽,全身骨骼肌发育异常,有先天性心脏病,这是死亡的重要原因。患儿智力有明显缺陷,但因存活时间很短,多数难以测量。帕陶氏综合征(13-三体)在新生儿中的发病率约为1:25000,女性明显多于男性。患儿的畸形和临床表现要比21三体严重得多,上唇裂,并常有腭裂,耳位低,脑和内脏的畸形非常普遍,智力发育障碍见于所有的患者,而且程度严重,存活较久的患儿还有癫痫样发作,肌张功力低下等。性染色体异常主要有xxy综合征,xyy综合征,turner综合征,超雌体。随着染色体分带技术、pcr技术、dna检测技术等的发展,对染色体畸变与疾病关系的认识日益加深,染色体病日趋增多。对于染色体异常的筛查与检测,有无创非侵入式和有创侵入式的方法,其中羊水穿刺是侵入式方法中最常见和准确的方法,用羊水细胞通过数字pcr技术可以快速准确的检测非整倍体。数字pcr(digitalpcr-dpcr)发展于1992年,sykes等使用有限稀释,pcr扩增,泊松分布校正的方法,通过判断终点信号的有或无,检测了复杂背景下低丰度的igh重链突变基因,进行了极其精细的定量研究。1997年,kalinina等使用毛细管作为反应容器,使用taqman探针收集到基因组dna的单分子信号,发展出了单分子定量技术。1999年vogelstein和kinzler提出了数字pcr的概念,主要是利用384孔板,检测结肠癌患者中的ras突变。2003年,dressman发明了beaming技术(磁珠,乳化,扩增,磁力),这也为后来的微滴式数字pcr(ddpcr)提供了技术基础。在此之后,数字pcr在商品化道路上演变成了扩增载体的发展。形成了后来的以life公司为代表的芯片式数字pcr系统(cdpcr)和bio-rad公司为代表的微滴式数字pcr系统(ddpcr)。数字pcr所做的就是把一个体系近于无限的稀释或者叫分割。将15ul的体系分割成20000份、10万份,分割到一个反应孔里面只有一条dna,孔中dna可能是野生型的,也可能是突变型的,也可能是无关的背景dna,最终实现了每个孔中只有一条单分子的模板,然后进行特异性扩增,只需要数出多少孔里有扩增,就知道目标基因的拷贝数。数字pcr技术是一种新的核酸检测和定量方法,与传统定量pcr(qpcr)技术不同,数字pcr采用绝对定量的方式,不依赖于标准曲线和参照样本,直接检测目标序列的拷贝数。由于这种检测方式具有比传统qpcr更加出色的灵敏度和特异性、精确性,dpcr迅速得到广泛的应用,这项技术在极微量核酸样本检测、复杂背景下稀有突变检测和表达量微小差异鉴定方面变现出的优势已被普遍认可。数字pcr基于单分子层面上的技术,可以摆脱qpcr对于标准品的依赖,使用pcr手段可以从过去的指数型衡量方式变为线性衡量。数字pcr的基本流程、原理如图1、2所示。目前,产前诊断主要分为侵入式的产前诊断和非侵入式的产前诊断,非侵入式的产前筛查虽然有早期快速无创的优势,但作为一种筛查手段,检测到的异常样本还应依赖羊膜穿刺进一步确定。根据华大基因2013年检出并得到核型分析验证的909例高风险样本中,无创产前诊断有6例t21假阳性和3例t18假阳性。主要原因可能是限制性胎盘嵌合(cpm)或胎儿为真正的嵌合体(tfm),这需要通过羊水或脐血染色体核型分析进一步明确诊断。核型分析作为产前检测的金标准,但在诊断胎儿染色体非整倍体实验周期较长,实验室需要经过一周的染色体培植来确定婴儿是否畸形,对操作人员的要求较高,而且存在检测失败的情况。技术实现要素:针对上述现有技术,本发明提供了数字pcr方法快速检测羊水细胞非整倍体,具体包括检测染色体非整倍体的扩增组合物,及快速检测试剂盒,以及检测方法。本发明是通过以下技术方案实现的:本发明利用生物信息学查找13/18/21/x/y染色体及1号染色体上特异的保守核酸序列,然后分别设计引物和taqman探针。1号染色体的特异高保守序列作为内参,13/18/21/x/y染色体的特异高保守序列作为目标基因,然后用数字pcr的方法检测对应染色体的拷贝数,经过比例计算得出对应染色体是否为非整倍体。检测染色体非整倍体的扩增组合物,包括以下的扩增引物和探针(序列方向均为5'to3'):针对1号染色体的内参基因上下游引物eif2c1-f,eif2c1-r:eif2c1-f:gttcggctttcaccagtc;eif2c1-r:ctccatagctctccccact;针对13-三体的目标基因上下游引物mbnl2-r,mbnl2-f:mbnl2-f:ctcacctatccacaatgca;mbnl2-r:gggattcaagcgaattaac;针对18-三体的目标基因上下游引物ehzf-r,ehzf-f:ehzf-f:ccagctggtacttggaaga;ehzf-r:tgtcgtatgtggagccaa;针对21-三体的目标基因上下游引物dscr4-f,dscr4-r:dscr4-f:ccctttccactcagaacca;dscr4-r:agcccatggaggtaaagtt;针对x染色体的目标基因上下游引物noncoding-f,noncoding-r:noncoding-f:gatgaggaaggcaatgat;noncoding-r:ttggcttttaccaaatag;针对y染色体的目标基因上下游引物sry-f,sry-r:sry-f:cgcttaacatagcagaagc;sry-r:agtttcgaactctggcac;6种taqman探针:eif2c1(chr1):cgccctgccatgtggaaga;mbnl2(chr13):aggtgcatcatgggaacgg;ehzf(chr18):tcagtgcctgcctggttcc;dscr4(chr21):cagcaattttgggcaaaatgt;noncoding(chrx):tgtttctctctgcctgcact;sry(chry):tgtcgcactctccttgtttttg。上述序列如表1所示,如seqidno.1~18所示。表1primer名称*序列(5'to3')*eif2c1-fgttcggctttcaccagtceif2c1-rctccatagctctccccacteif2c1-taqmanprobecgccctgccatgtggaagambnl2-fctcacctatccacaatgcambnl2-rgggattcaagcgaattaacmbnl2-taqmanprobeaggtgcatcatgggaacggehzf-fccagctggtacttggaagaehzf-rtgtcgtatgtggagccaaehzf-taqmanprobetcagtgcctgcctggttccdscr4-fccctttccactcagaaccadscr4-ragcccatggaggtaaagttdscr4-taqmanprobecagcaattttgggcaaaatgtnoncoding-fgatgaggaaggcaatgatnoncoding-rttggcttttaccaaatagnoncoding--taqmanprobetgtttctctctgcctgcactsry-fcgcttaacatagcagaagcsry-ragtttcgaactctggcacsry-taqmanprobetgtcgcactctccttgtttttg进一步地,所述taqman探针,采用vic或fam荧光标记。更进一步地,荧光标记具体如下:eif2c1(chr1),vic标记;mbnl2(chr13),fam标记;ehzf(chr18),fam标记;dscr4(chr21),fam标记;noncoding(chrx),fam标记;sry(chry),fam标记。检测染色体非整倍体的检测试剂盒,包括上述检测染色体非整倍体的扩增组合物,以及用于进行数字pcr的反应体系,包括:pcr反应缓冲液、dntp混合物、羊水dna模板、taq酶和ddh2o。检测染色体非整倍体的方法,包括以下步骤:(一)提取羊水样本dna;(二)数字pcr检测羊水dna非整倍体:利用数字pcr检测芯片每个反应单元的fam和vic荧光,分别计算13、18、21、x、y染色体的拷贝数。进一步地,所述步骤(一)提取羊水样本dna的具体方式如下:(1)将羊水进行4℃,3000rcf离心15min,弃上清,保留250μl上清液和全部沉淀物(根据沉淀物多少进行保留,沉淀物较多就保留250μl,少则保留200μl),混匀后将其转移至新的1.5ml低吸附管中;(2)加入20μlproteinasek,充分涡旋混匀,瞬离,加200μlbufferal溶液,涡旋混匀20s,短暂离心,56℃温浴10min;(3)向上述1.5ml低吸附管中加入200μl的无水乙醇,涡旋混匀15s,短暂离心5s;(4)取出吸附柱,并根据样本信息在吸附柱管盖上标记样本编号;(5)将离心管中的溶液全部转至吸附柱中(注意:转移样本时不要把样本加到吸附柱内壁),8000rpm离心1min,弃废液,将吸附柱管转移至新的收集管中;(6)向上述的吸附柱内加入500μlbufferaw1(使用前检查是否已加入相应的无水乙醇),12000rpm离心1min,弃废液,将吸附柱转移至新的收集管中;(7)向吸附柱中加入500μlbufferaw2(使用前检查是否已加入相应的无水乙醇),14000rpm离心3min,弃废液,将吸附柱转移至新的收集管中;(8)将空吸附柱14000rpm离心3min,用10μl枪头吸走残留液体,将吸附柱转移至新的1.5ml低吸附管中,打开吸附盖子,室温晾干3min;(9)向上述的吸附柱的硅胶膜正上方悬空加入50μlbufferae,室温放置5min,14000rpm离心3min;(10)将收集管中的液体全部重新转移至所对应的吸附柱的硅胶膜上,室温放置5min,14000rpm离心3min(浓度不够的情况下可以重新洗);(11)离心后须核对吸附柱编号和低吸附管管盖上编号无误,并确认1.5ml低吸附管中有dna溶液后方可把吸附柱丢掉;(12)使用nanodrop2000分光光度计对所得的dna样本进行浓度的测定,-20℃保存备用。进一步地,所述步骤(二)数字pcr检测羊水dna非整倍体的具体方式如下:(1)提前将试剂取出,室温融化,短暂震荡,瞬时离心;按如下体系分别配置:vic探针-引物mix:引物终浓度13.5um,探针终浓度3um(15ul体系中,1ul中引物为900nm,探针为200nm);fam探针-引物mix:引物终浓度13.5um,探针终浓度3um(15ul体系中,1ul中引物为900nm,探针为200nm);将内参基因vic探针-引物mix分别与五种目标基因fam探针-引物mix等体积混合备用;(2)dpcr体系配制:quantstudio3ddigitalpcrmastermixv2(2x)7.5μl,探针-引物mix2μl,羊水dna模板50ng,补足水至15μl;(3)上样:将14.5μl反应预混液加到上样拨片的加样孔中,随后下压按钮,加样器会自动载入并将反应预混液均匀分散到两万个芯片反应孔中,当上样臂返回起始位点时,用足量的浸没液完整覆盖整个芯片表面;pcr扩增反应程序:96℃,10min;(60℃2min,98℃30s)44cycle;60℃2min;10℃保存。(4)数据读取:插入芯片,自动读取数据,分析结束后,查看数据计算vic/fam比值;当vic/fam比值等于或约等于2:0时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:0,无目标染色体;当vic/fam比值等于或约等于1:1时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于1:1,目标染色体为二倍体,样本正常;当vic/fam比值等于或约等于2:1时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:1,目标染色体为单倍体;当vic/fam比值等于或约等于2:3时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:3,目标染色体为三倍体。本发明利用数字pcr技术对正常妊娠妇女进行产前检测,通过常染色体与目标染色体(13,18,21,x,y)之间计数的差异确定目标染色体的倍性情况。采用本发明的数字pcr的方法检测羊水中胎儿染色体非整倍体,最快可在7个小时内完成实验,大大缩短了实验周期,对于无创产前筛查中的阳性或假阳性样本都可以进行快速的验证,减少了患者家属焦急等待的时间,而且准确性提高,不存在检测失败的情况,在一定程度上降低了成本。附图说明图1:数字pcr的基本流程,包括样本的稀释,dpcr体系的配置,dpcr的上样与分割,dpcr的扩增,dpcr结果分析。图2:数字pcr的原理。图3:应用实例1中芯片1、2、3、4、5的数字pcr扩增芯片结果示意图及统计结果图。图示:芯片1、2、3、4、5分别代表通过数字pcr方法检测样本a的13、18、21、x、y非整倍体扩增结果示意图,fam荧光信号值代表单孔中只有目标基因的扩增数目,vic荧光信号值代表单孔中只有内参基因的扩增数目,fam+vic荧光信号值代表单孔中有目标基因和内参基因的扩增数目;6代表样本a的13、18、21、x、y非整倍体检测结果统计图,即目标基因拷贝数在总拷贝数中的占比。可知样本a是13、18、21倍性正常的女孩。图4:应用实例2中芯片7、8、9、10、11的数字pcr扩增芯片结果示意图及统计结果图。图示:芯片7、8、9、10、11分别代表通过数字pcr方法检测样本b的13、18、21、x、y非整倍体扩增结果示意图,fam荧光信号值代表单孔中只有目标基因的扩增数目,vic荧光信号值代表单孔中只有内参基因的扩增数目,fam+vic荧光信号值代表单孔中有目标基因和内参基因的扩增数目;12代表样本b的13、18、21、x、y非整倍体检测结果统计图,即目标基因拷贝数在总拷贝数中的占比。可知样本b是13、18倍性正常,21-三体的男孩。图5:应用实例3中芯片13、14、15、16、17的数字pcr扩增芯片结果示意图及统计结果图。图示:13、14、15、16、17分别代表通过数字pcr方法检测样本c的13、18、21、x、y非整倍体扩增结果示意图,fam荧光信号值代表单孔中只有目标基因的扩增数目,vic荧光信号值代表单孔中只有内参基因的扩增数目,fam+vic荧光信号值代表单孔中有目标基因和内参基因的扩增数目;18代表样本c的13、18、21、x、y非整倍体检测结果统计图,即目标基因拷贝数在总拷贝数中的占比。可知样本c是13、18倍性正常,21-三体的女孩。具体实施方式下面结合实施例对本发明作进一步的说明。下述实施例中所涉及的仪器、试剂、材料等,若无特别说明,均为现有技术中已有的常规仪器、试剂、材料等,可通过正规商业途径获得。下述实施例中所涉及的实验方法,检测方法等,若无特别说明,均为现有技术中已有的常规实验方法,检测方法等。下述实施例中,数字pcr实验试剂及设备购于abi公司,包括quantstudio3ddigitalchipkitv2,quantstudio3ddigitalpcrmastermixv2;引物及探针由invitrogen公司合成。实施例1检测染色体非整倍体的扩增组合物、检测试剂盒及方法利用生物信息学查找13/18/21/x/y染色体及1号染色体上特异的保守核酸序列,然后分别设计引物和taqman探针。1号染色体的特异高保守序列作为内参,13/18/21/x/y染色体的特异高保守序列作为目标基因,然后用数字pcr的方法检测对应染色体的拷贝数,经过比例计算得出对应染色体是否为非整倍体。检测染色体非整倍体的扩增组合物,包括以下的扩增引物和探针(序列方向均为5'to3'):针对1号染色体的内参基因上下游引物eif2c1-f,eif2c1-r:eif2c1-f:gttcggctttcaccagtc;eif2c1-r:ctccatagctctccccact;针对13-三体的目标基因上下游引物mbnl2-r,mbnl2-f:mbnl2-f:ctcacctatccacaatgca;mbnl2-r:gggattcaagcgaattaac;针对18-三体的目标基因上下游引物ehzf-r,ehzf-f:ehzf-f:ccagctggtacttggaaga;ehzf-r:tgtcgtatgtggagccaa;针对21-三体的目标基因上下游引物dscr4-f,dscr4-r:dscr4-f:ccctttccactcagaacca;dscr4-r:agcccatggaggtaaagtt;针对x染色体的目标基因上下游引物noncoding-f,noncoding-r:noncoding-f:gatgaggaaggcaatgat;noncoding-r:ttggcttttaccaaatag;针对y染色体的目标基因上下游引物sry-f,sry-r:sry-f:cgcttaacatagcagaagc;sry-r:agtttcgaactctggcac;6种taqman探针:eif2c1(chr1):cgccctgccatgtggaaga;mbnl2(chr13):aggtgcatcatgggaacgg;ehzf(chr18):tcagtgcctgcctggttcc;dscr4(chr21):cagcaattttgggcaaaatgt;noncoding(chrx):tgtttctctctgcctgcact;sry(chry):tgtcgcactctccttgtttttg。上述序列如表1所示,如seqidno.1~18所示。荧光标记具体如下:eif2c1(chr1),vic标记;mbnl2(chr13),fam标记;ehzf(chr18),fam标记;dscr4(chr21),fam标记;noncoding(chrx),fam标记;sry(chry),fam标记。检测染色体非整倍体的检测试剂盒,包括上述检测染色体非整倍体的扩增组合物,以及用于进行数字pcr的反应体系,包括:pcr反应缓冲液、dntp混合物、羊水dna模板、taq酶和ddh2o。检测染色体非整倍体的方法,包括以下步骤:(一)提取羊水样本dna;(二)数字pcr检测羊水dna非整倍体:利用数字pcr检测芯片每个反应单元的fam和vic荧光,分别计算13、18、21、x、y染色体的拷贝数。所述步骤(一)提取羊水样本dna的具体方式如下:(1)将羊水进行4℃,3000rcf离心15min,弃上清,保留250μl上清液和全部沉淀物(根据沉淀物多少进行保留,沉淀物较多就保留250μl,少则保留200μl),混匀后将其转移至新的1.5ml低吸附管中;(2)加入20μlproteinasek,充分涡旋混匀,瞬离,加200μlbufferal溶液,涡旋混匀20s,短暂离心,56℃温浴10min;(3)向上述1.5ml低吸附管中加入200μl的无水乙醇,涡旋混匀15s,短暂离心5s;(4)取出吸附柱,并根据样本信息在吸附柱管盖上标记样本编号;(5)将离心管中的溶液全部转至吸附柱中(注意:转移样本时不要把样本加到吸附柱内壁),8000rpm离心1min,弃废液,将吸附柱管转移至新的收集管中;(6)向上述的吸附柱内加入500μlbufferaw1(使用前检查是否已加入相应的无水乙醇),12000rpm离心1min,弃废液,将吸附柱转移至新的收集管中;(7)向吸附柱中加入500μlbufferaw2(使用前检查是否已加入相应的无水乙醇),14000rpm离心3min,弃废液,将吸附柱转移至新的收集管中;(8)将空吸附柱14000rpm离心3min,用10μl枪头吸走残留液体,将吸附柱转移至新的1.5ml低吸附管中,打开吸附盖子,室温晾干3min;(9)向上述的吸附柱的硅胶膜正上方悬空加入50μlbufferae,室温放置5min,14000rpm离心3min;(10)将收集管中的液体全部重新转移至所对应的吸附柱的硅胶膜上,室温放置5min,14000rpm离心3min(浓度不够的情况下可以重新洗);(11)离心后须核对吸附柱编号和低吸附管管盖上编号无误,并确认1.5ml低吸附管中有dna溶液后方可把吸附柱丢掉;(12)使用nanodrop2000分光光度计对所得的dna样本进行浓度的测定,-20℃保存备用。所述步骤(二)数字pcr检测羊水dna非整倍体的具体方式如下:(1)提前将试剂取出,室温融化,短暂震荡,瞬时离心;按如下体系分别配置:vic探针-引物mix:引物终浓度13.5um,探针终浓度3um(15ul体系中,1ul中引物为900nm,探针为200nm);fam探针-引物mix:引物终浓度13.5um,探针终浓度3um(15ul体系中,1ul中引物为900nm,探针为200nm);将内参基因vic探针-引物mix分别与五种目标基因fam探针-引物mix等体积混合备用;(2)dpcr体系配制:quantstudio3ddigitalpcrmastermixv2(2x)7.5μl,探针-引物mix2μl,羊水dna模板50ng,补足水至15μl;(3)上样:将14.5μl反应预混液加到上样拨片的加样孔中,随后下压按钮,加样器会自动载入并将反应预混液均匀分散到两万个芯片反应孔中,当上样臂返回起始位点时,用足量的浸没液完整覆盖整个芯片表面;pcr扩增反应程序:96℃,10min;(60℃2min,98℃30s)44cycle;60℃2min;10℃保存。(4)数据读取:插入芯片,自动读取数据,分析结束后,查看数据计算vic/fam比值;当vic/fam比值等于或约等于2:0时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:0,无目标染色体;当vic/fam比值等于或约等于1:1时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于1:1,目标染色体为二倍体,样本正常;当vic/fam比值等于或约等于2:1时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:1,目标染色体为单倍体;当vic/fam比值等于或约等于2:3时,则内参基因拷贝数/目标基因拷贝数比例等于或约等于2:3,目标染色体为三倍体。应用实例1:应用数字pcr技术对样本a进行13,18,21,x,y非整倍体检测步骤如下:1.取孕妇羊水样本a通过吸附柱法提取dna。2.分别配置5个数字pcr反应混合液,将羊水样本a稀释到50ng,取1ul加入其中。3.通过数字pcr上样系统将五个混合体系上样,分割到五张芯片中。4.通过数字pcr扩增仪进行扩增。5.将五张芯片放入数字pcr分析系统中读取结果,如图3所示。6.图3结果显示:芯片1中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=51.327%,即13号染色体:1号染色体比例约为1:1,13号染色体为二倍体,倍性正常。芯片2中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=49.502%,即18号染色体:1号染色体比例约为1:1,18号染色体为二倍体,倍性正常。芯片3中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=50.54%,即21号染色体:1号染色体比例约为1:1,21号染色体为二倍体,倍性正常。芯片4中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=49.502%,即x染色体:1号染色体比例约为1:1,x染色体为二倍体,倍性正常。芯片5中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=1.49e-2%,即y染色体:1号染色体比例约为0:2,无y染色体。样本a是13、18、21倍性正常的女孩。应用实例2:应用数字pcr技术对样本b进行13,18,21,x,y非整倍体检测步骤如下:1.取孕妇羊水样本b通过吸附柱法提取dna。2.分别配置5个数字pcr反应混合液,将羊水样本a稀释到50ng,取1ul加入其中。3.通过数字pcr上样系统将五个混合体系上样,分割到五张芯片中。4.通过数字pcr扩增仪进行扩增。5.将五张芯片放入数字pcr分析系统中读取结果,如图4所示。6.图4结果显示:芯片7中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=49.68%,即13号染色体:1号染色体比例约为1:1,13号染色体为二倍体,倍性正常。芯片8中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=51.043%,即18号染色体:1号染色体比例约为1:1,18号染色体为二倍体,倍性正常。芯片9中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=62.204%,即21号染色体:1号染色体比例约为3:2,21号染色体为三倍体,样本b为21-三体。芯片10中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=35.367%,即x染色体:1号染色体比例约为1:2,x染色体为单倍体。芯片11中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=34.953%,即y染色体:1号染色体比例约为1:2,y染色体为单倍体。样本b是13、18倍性正常,21-三体的男孩。应用实例3:应用数字pcr技术对样本c进行13,18,21,x,y非整倍体检测步骤如下:1.取孕妇羊水样本c通过吸附柱法提取dna。2.分别配置5个数字pcr反应混合液,将羊水样本c稀释到50ng,取1ul加入其中。3.通过数字pcr上样系统将五个混合体系上样,分割到五张芯片中。4.通过数字pcr扩增仪进行扩增。5.将五张芯片放入数字pcr分析系统中读取结果,如图5所示。6.图5结果显示:芯片13中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=50.213%,即13号染色体:1号染色体比例约为1:1,13号染色体为二倍体,倍性正常。芯片14中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=52.791%,即18号染色体:1号染色体比例约为1:1,18号染色体为二倍体,倍性正常。芯片15中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=60.663%,即21号染色体:1号染色体比例约为3:2,21号染色体为三倍体,样本c为21-三体。芯片16中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=49.708%,即x染色体:1号染色体比例约为1:1,x染色体为二倍体,倍性正常。芯片17中,目标基因拷贝数/(目标基因拷贝数+内参基因拷贝数)=0.132%,即y染色体:1号染色体比例约为0:2,无y染色体。样本c是13、18倍性正常,21-三体的女孩。通过上述的应用实例,可以进一步说明,本发明具有以下优点:(1)试验周期短:检测羊水中胎儿染色体非整倍体最快可在7个小时内完成实验,对于无创产前筛查中的阳性或假阳性样本都可以进行快速的验证,结果以图谱的形式展示,不需要进行繁琐的分析,方便读取。(2)准确性高:因为是染色体疾病的筛查,不会因为个体的生理变化而改变检测结果,对于同一个样本,样本dna浓度的高低都不会影响实验最终比例,只有拷贝数高低之分。通过进一步优化实验体系,发现dna浓度为50ng时染色体拷贝数可以达到最优。通过高分辨率染色体示意图,可以分辨出每一张芯片fam与vic的荧光信号及具体值,如对于fam标记的sry基因因为只存在与y染色体上,高分辨率图谱显示几乎没有fam的荧光信号时,可准确判断出样本为女孩。在如当样本为男性样本时,fam荧光信号数目在高分辨率染色体示意图中占vic信号的一半。通过荧光信号数目来判断染色体异常及胎儿性别显然是高效准确的。进一步进行泊松统计,可精确得到目标基因和内参基因每微升的拷贝数及比例,佐证之前的判断结果。大量的实验证明通过本发明的方法检测羊水细胞的13,18,21,x,y非整倍体是快速准确的。给本领域技术人员提供上述实施例,以完全公开和描述如何实施和使用所主张的实施方案,而不是用于限制本文公开的范围。对于本领域技术人员而言显而易见的修饰将在所附权利要求的范围内。本说明书引述的所有出版物、专利和专利申请通过引用并入本文,如同这些出版物、专利和专利申请各自特别地和个别地表明通过引用并入本文。序列表<110>银丰基因科技有限公司银丰生物工程集团有限公司<120>数字pcr方法快速检测羊水细胞非整倍体<141>2017-12-26<160>18<170>siposequencelisting1.0<210>1<211>18<212>dna<213>artificialsequence<400>1gttcggctttcaccagtc18<210>2<211>19<212>dna<213>artificialsequence<400>2ctccatagctctccccact19<210>3<211>19<212>dna<213>artificialsequence<400>3cgccctgccatgtggaaga19<210>4<211>19<212>dna<213>artificialsequence<400>4ctcacctatccacaatgca19<210>5<211>19<212>dna<213>artificialsequence<400>5gggattcaagcgaattaac19<210>6<211>19<212>dna<213>artificialsequence<400>6aggtgcatcatgggaacgg19<210>7<211>19<212>dna<213>artificialsequence<400>7ccagctggtacttggaaga19<210>8<211>18<212>dna<213>artificialsequence<400>8tgtcgtatgtggagccaa18<210>9<211>19<212>dna<213>artificialsequence<400>9tcagtgcctgcctggttcc19<210>10<211>19<212>dna<213>artificialsequence<400>10ccctttccactcagaacca19<210>11<211>19<212>dna<213>artificialsequence<400>11agcccatggaggtaaagtt19<210>12<211>21<212>dna<213>artificialsequence<400>12cagcaattttgggcaaaatgt21<210>13<211>18<212>dna<213>artificialsequence<400>13gatgaggaaggcaatgat18<210>14<211>18<212>dna<213>artificialsequence<400>14ttggcttttaccaaatag18<210>15<211>20<212>dna<213>artificialsequence<400>15tgtttctctctgcctgcact20<210>16<211>19<212>dna<213>artificialsequence<400>16cgcttaacatagcagaagc19<210>17<211>18<212>dna<213>artificialsequence<400>17agtttcgaactctggcac18<210>18<211>22<212>dna<213>artificialsequence<400>18tgtcgcactctccttgtttttg22当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1