用于保护聚合物对抗紫外光的添加剂的制作方法
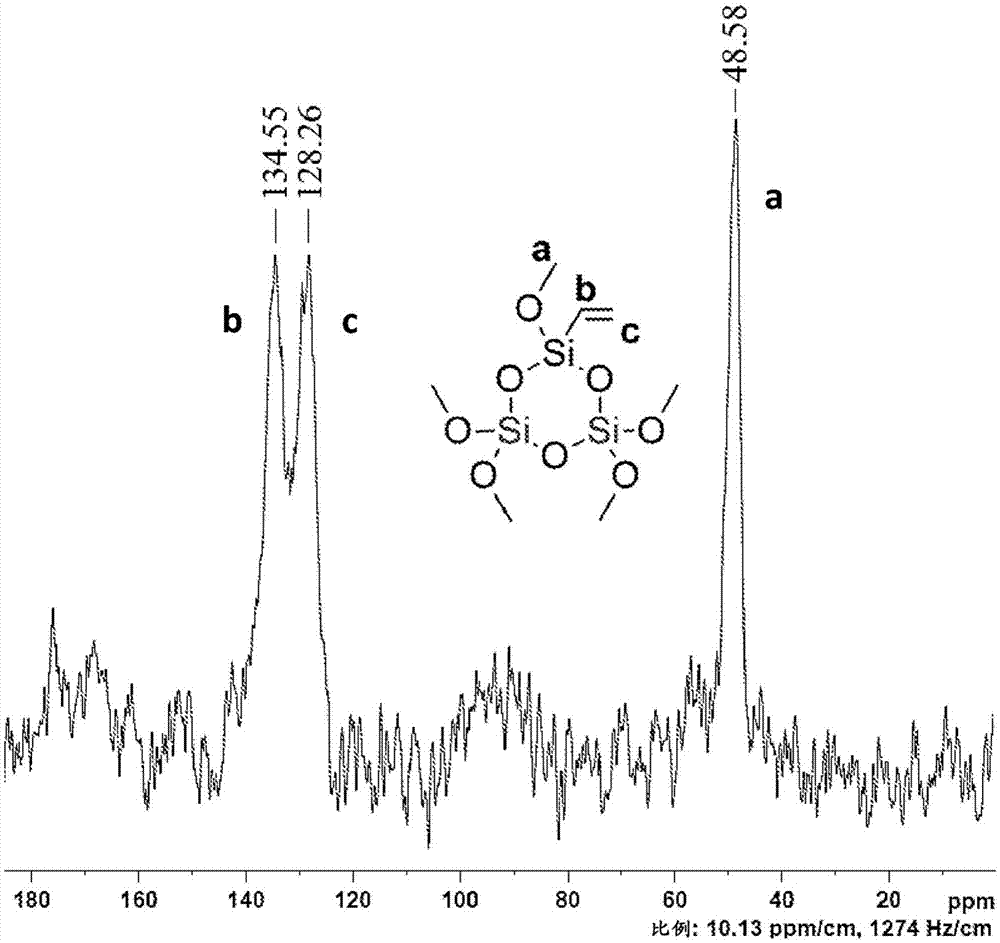
一些添加剂倾向于与本体聚合物分离并且迁移到聚合物表面。出于该原因,必须在严格的储存条件下测试聚合物组合物以测量该表面迁移的发生。添加剂的表面迁移在食品包装材料中是特别不可接受的。因此,已经提出将意在用作食品包装材料中的添加剂的紫外光吸收剂(uva)进行化学改性以增加其尺寸,努力减少它们向表面的迁移。另一方法包括在结构上改性包装材料(如jp2013075453和cn202784136中所示),例如,通过使用由多个层组成的层合包装材料,其具有在含uva的层的顶部上施加的不含uva的层。现已发现,紫外光吸收化合物可以与无机氧化物颗粒(例如二氧化硅、氧化锌)的表面共价结合;任选地借助于适宜的间隔基基团进行共价结合,并且如此形成的颗粒可以与各种聚合物混配以形成颗粒,特别是聚合物膜,从而显示出uv吸收剂的非常低的表面迁移。例如,为支持本发明进行的实验工作表明,引入添加剂的聚丙烯和线性低密度聚乙烯(lldpe)膜能够随时间推移保持基本恒定的uv阻挡水平,而在测试期间没有检测到紫外光吸收化合物的表面迁移。已经报道了活性分子(包括光吸收有机化合物)与二氧化硅颗粒表面的共价结合(us6,685,924,名称为merck),目的是形成用于皮肤病制剂和化妆品制剂的缀合物。示例性缀合物具有以下结构:us2008/0260664描述了用有机化合物(包括光吸收化合物)官能化的无机颗粒(例如二氧化硅)。有机化合物主要存在于颗粒的核中。现已发现,紫外光吸收化合物(例如,苯并三唑和二苯甲酮类)可以如下共价连接到无机氧化物颗粒的表面:或者对颗粒进行依序表面改性,然后使改性表面与紫外光吸收化合物(本申请称为“依序表面改性变体”或“第一变体”)反应;或者使紫外光吸收化合物与未改性的无机氧化物颗粒(本申请称为“一步变体”或“第二变体”)直接反应。在任何情况下,在其表面上具有紫外光吸收化合物的所得颗粒可用作添加剂以修改聚合物商业用途中的性质,即改善光稳定性。因此,本发明主要涉及适宜用作聚合物中的添加剂的紫外光吸收剂,其包含无机氧化物颗粒,所述颗粒在其表面上具有紫外光吸收化合物,其中所述紫外光吸收化合物或与所述颗粒表面上提供的间隔基化学(共价)键合、或直接键合到颗粒表面。无机氧化物优选地选自二氧化硅和氧化锌。关于第一变体,二氧化硅颗粒由于羟基(或者更确切地说,其表面上的硅烷醇基团(sioh))的存在而具有表面反应性,可用于化学键形成。优选地,在二氧化硅表面上构建有机硅间隔基,以使表面和紫外光吸收化合物之间能够连接。间隔基可以通过一系列化学反应构建在二氧化硅表面上以提供官能团,该官能团进而可以与紫外光吸收化合物反应。例如,二氧化硅颗粒的表面通过引入sih基团来改性。氢化硅键与多种化学官能团(烯烃、醇、醛、酮)的高化学反应性允许将适当官能化的紫外光吸收化合物连接到二氧化硅颗粒上。例如,在二氧化硅表面上形成的氢化硅基团可以加成到具有紫外光吸收部分的烯属化合物的碳-碳双键中。适用于本发明第一变体的二氧化硅优选为热解法二氧化硅。对于筛选用于本发明第一变体中的热解法二氧化硅重要的性质包括二氧化硅上的硅烷醇基团的粒度、比表面积和浓度。因此,在本发明的该变体中有用的二氧化硅是热解法二氧化硅,其颗粒的平均大小为小于40nm,例如为5nm至35nm,更具体地为5nm至20nm,甚至更具体地为5nm至15nm,其中比表面积为不小于150m2/g,例如不小于200m2/g,例如200至500m2/g。二氧化硅表面上的sioh基团浓度是优选不小于0.7mmol/g,例如不小于0.8mmol/g,例如0.8mmol/g至3.0mmol/g(通过热重分析测量)。可以使用商购可得的热解法二氧化硅等级如来自cabotcorporation的m5和eh5,以及来自evonikindustries的200和300/30。关于第二变体,可以使用二氧化硅或氧化锌颗粒,也就是说,本发明的这种变体包括使无机氧化物颗粒直接与具有适宜反应性基团如si-o-烷基基团的紫外光吸收化合物反应。用于该'一步变体'的二氧化硅包括具有上述性质的热解法二氧化硅,例如,m5和eh5。在下面报道的实验工作中测试的另一种类型的二氧化硅是胶体二氧化硅,即悬浮在溶剂中的二氧化硅。由一群离散、球形的均匀大小的颗粒组成的单分散胶体二氧化硅可以在最终聚合物产品中显示出优势,这归因于其能够在聚合物产品中产生均匀的添加剂分布。如下面报道的实验工作所示,我们能够找到允许这种类型的胶体二氧化硅与紫外光吸收化合物在下述溶剂中迅速反应的反应条件,所述溶剂与市场上可获得的商业产品中用于悬浮胶体二氧化硅的溶剂相同。现由此描述'依序表面改性变体'和'一步变体'。二氧化硅颗粒的依序表面改性包括在二氧化硅表面上引入第一官能团,然后与紫外光吸收化合物反应;或将第一官能团引入到二氧化硅表面上,然后将所述第一官能团转化为多个第二官能团,所述第二官能团进而与紫外光吸收化合物反应。第二种方法通常是优选的,因为每个单独的第一官能团与带有多个第二官能团的分子反应,导致最终可用于与紫外光吸收化合物形成化学键的反应性位点的浓度增加。因此,本发明主要涉及制备用于保护聚合物免受紫外辐射的紫外光吸收剂添加剂的方法,其包括在无机氧化物颗粒的表面上引入第一官能团,使由所述第一基团改性的如此形成的无机颗粒与具有多个第二官能团的间隔基反应;和使由所述第二官能团改性的如此形成的无机颗粒与紫外光吸收化合物反应。为此,在二氧化硅表面上如下引入第一官能团(优选碳-碳双键):将二氧化硅颗粒与乙烯基化合物反应,所述乙烯基化合物例如为式(h2c=ch)isi(ocjh2j+1)4-i的乙烯基化合物,例如,乙烯基三甲氧基硅烷(i=1,j=1)或三乙烯基乙氧基硅烷(i=3,j=2)。这两种乙烯基化合物是高沸点液体,反应可以如下进行:在不含溶剂的反应介质中,使用过量的(h2c=ch)isi(ocjh2j+1)4-i以提供可搅拌的反应混合物。因此,向反应容器中加入二氧化硅颗粒和乙烯基化合物,并且在加热下(例如在高于100℃,优选在回流温度)在搅拌下进行反应,得到带有乙烯基的二氧化硅颗粒。几小时后使反应完全。冷却反应混合物,通过过滤分离表面改性的颗粒,洗涤除去过量的反应物,并且干燥。接着,带有多个官能团的间隔基与存在于二氧化硅颗粒表面上的第一官能团化学连接。间隔基中官能团的数量由n表示,其中n是等于或大于3的整数。应该注意的是,这n个官能团中的一个参与与二氧化硅颗粒表面上存在的第一官能团的键形成反应,因此保留至多n-1个可用于与相应n-1个紫外光吸收化合物的键形成反应的反应位点。间隔基中官能团的数量优选不小于四个(n≥4)。优选地,间隔基具有多个彼此相同的官能团,例如多个氢化硅基团。氢化硅官能团具有两个有用的特征。首先,在适宜的催化剂存在下,氢化硅与碳-碳双键的加成反应(氢化硅烷化)迅速进行。此外,如上所述,待置于二氧化硅表面上的氢化硅基团与各种化学官能团的高化学反应性,使得许多不同的紫外光吸收化合物的连接在添加剂形成的最后步骤中成为可能。一些示例性的包含多个氢化硅基团的有机硅间隔基如下所示:(n=4;下文中为"a4")(n=6;下文中为"a6")(n=6;下文中为"a6y")(n≥10;例如n=75)应该记住,带有紫外光吸收化合物的二氧化硅意在用作聚合物中的添加剂。因此,可以加载到二氧化硅颗粒表面上的紫外光吸收化合物的浓度越高,最终聚合物制剂中最终需要的二氧化硅添加剂的量就越低。因此,影响紫外光吸收化合物在二氧化硅表面上可达到的浓度的一个主要因素是所用间隔基中的官能团数量。筛选适宜间隔基的另一重要性质是二氧化硅-间隔基-uva添加剂与相关聚合物制剂的相容性。优选地,间隔基分子中的官能团数量是4个至80个(4≤n≤80)。本发明中可操作的一类间隔基分子的特征在于间隔基分子中的官能团数量是4个至10个(包括端值)(4≤n≤10)。对于4≤n≤10,体积大的间隔基优于‘线性’间隔基,也就是说,其中氢化硅基团是侧基并且氢化硅基团中硅原子的最近环境由两个烃基如烷基(例如甲基、乙基)或苯基占据的化合物,优于其中氢化硅是主链的一部分的化合物。下面报道的实验结果表明,在用“体积大的”a6y间隔基代替线性a6间隔基时,二氧化硅添加剂中紫外光吸收化合物的浓度几乎增加了两倍(0.3mmol/g至0.5mmol/g)。本发明中可操作的另一类间隔基分子的特征在于间隔基分子中的官能团数量大于20个(n≥20)。这些间隔基分子具有长的(n≥20)主链,其中氢化硅基团是重复单元的一部分;特别地,由下面报道的研究得出,由10个至80个重复单元组成的聚(甲基氢硅氧烷)作为优异的间隔基。例如,已显示由约50-80(例如约75)个氢化硅基团组成且优选用三甲基甲硅烷氧基封端的聚(甲基氢硅氧烷)使二氧化硅表面上的紫外光吸收化合物的浓度增至约2mmol/g。适宜用作本发明中的间隔基的聚(甲基氢硅氧烷)商购可得,例如,mh30(下文中为mh30;来自abspecialtysilicones)。实验结果也表明,紫外光吸收化合物与mh30的化学键合导致硅氧烷的热稳定性增强。间隔基与在二氧化硅表面上提供的乙烯基官能团的加成在溶剂中进行。溶剂优选为芳族烃,例如烷基取代的芳族烃如甲苯。反应在加热下、例如在温度为40℃至90℃进行。借助于催化剂促进加成反应,所述催化剂优选来自第viii族金属,特别是铂。选择催化剂使反应能够在低得足以使副反应最小化的温度平稳地进行。例如,当带有氢化硅基团的间隔基是甲基氢聚硅氧烷时,则反应温度不应超过60℃,例如约50℃。与上述间隔基的其它加成反应允许在较高温度(例如约70-80℃)在较短反应时间(例如小于10小时)进行。示例性pt催化剂包括[(c2h5)2s]2ptcl2,其证明在将含有氢化硅的间隔基加成至乙烯基改性的二氧化硅中非常有用。也可以使用pt2[(me2sich=ch2)2o]3,其命名为karstedt催化剂,或来自umicore的商购可得催化剂:umicorehs425或umicorehs432。也可以使用其它含过渡金属的催化剂,如基于铑或钌的催化剂,例如[cp*ru(mecn)3]pf6。在选定的升高温度在搅拌下维持反应混合物。完成加成反应后,通过过滤将颗粒(即,氢化硅改性的二氧化硅)从反应混合物中分离,并且任选地洗涤和干燥。接着,使氢化硅改性的二氧化硅与紫外光吸收化合物反应。例如,已知苯并三唑类的有机化合物吸收300nm至400nm范围内的紫外光,并且可用于本发明。优选苯并三唑,其中苯并三唑稠合体系的2位上的氮原子与取代的苯环键合。苯基部分上的取代基优选包括羟基。羟基苯基-苯并三唑衍生物在众多聚合物中显示出有效的紫外光保护。因此,更具体地,用于本发明的优选的紫外光吸收化合物是2-(2’-羟基苯基)苯并三唑衍生物:其中苯环进一步由反应性官能团取代。特别优选2-(2’-羟基苯基)苯并三唑衍生物,其中2’-羟基苯基环由显示出与氢化硅的高反应性的官能团如乙烯基或烯丙基取代。例如,2-(3-烯丙基-2-羟基-5-甲基-苯基)-2h-苯并三唑[也称为2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚]具有下述结构:其通过间隔基的氢化硅基团加成至烯丙基的碳-碳双键从而化学键合至二氧化硅颗粒。因此,在适宜的催化剂(例如,如上所述pt催化剂)存在下,将氢化硅表面改性的二氧化硅颗粒和2-(2'-羟基苯基)苯并三唑紫外光吸收化合物在有机溶剂中组合在一起。例如,将催化剂加入到预先加入在芳族烃中的二氧化硅和2-(2'-羟基苯基)苯并三唑衍生物的反应容器中,并且将反应混合物在搅拌下保持加热以完成加成反应。将由化学连接紫外光吸收化合物的二氧化硅颗粒组成的产物通过过滤分离、洗涤并干燥。收集滤液以从中回收未反应的紫外光吸收化合物。“依序表面改性变体”在其最优选的实施方案中产生适宜用作聚合物中的添加剂的光稳定剂。该光稳定剂形成本发明的具体实施方案;其包括在颗粒表面上具有紫外光吸收化合物的二氧化硅颗粒,其中紫外光吸收化合物的碳原子与有机硅间隔基的硅原子共价键合,其特征在于除了所述碳原子外,所述硅原子的最近环境还包括一个氧原子和两个烃基或包括两个氧原子和一个烃基。烃基包括但不限于烷基(如甲基或乙基)和苯基。更具体地,uva光吸收化合物是2-(2’-羟基苯基)苯并三唑化合物,其通过亚烷基链(-ch2-)n(n=2-5,例如n=3)共价键合至有机硅间隔基的硅原子,所述亚烷基链与所述羟基苯基-苯并三唑化合物的苯环连接。优选地,二氧化硅中的紫外光吸收化合物的浓度为不小于0.07mmol/g,例如不小于0.15mmol/g,更优选不小于0.2mmol/g,例如0.2至3.0mmol/g,例如0.3至0.7mmol/g或2.0mmol/g至2.5mmol/g(通过下面示例的tga测量)。添加剂也可包含一些氢化硅基团。现涉及‘一步变体’,它基于紫外光吸收化合物与未改性的二氧化硅颗粒或氧化锌颗粒的直接反应。具有能够与表面-oh基团进行键形成反应的官能团的任何紫外光吸收化合物可用于‘一步变体’,如取代有-si(-o-烷基)3例如-si(-o-c2h5)3的紫外光吸收化合物。下面描述两种带有-si(-o-c2h5)3官能团的紫外光吸收化合物,一种是2-羟基二苯甲酮类,另一种是2-(2'-羟基苯基)苯并三唑类:[以上描述的2’-羟基苯基)苯并三唑由farkas等人在molecules(15),p.6205-6216(2010)中描述]因此,无机氧化物如二氧化硅或氧化锌与紫外光吸收化合物在适宜的有机溶剂中(例如在芳族烃如甲苯中)在搅拌和加热下优选在溶剂沸点在回流下混合。完成后,冷却反应混合物并将产物与液相分离、洗涤和干燥。或者,用商购可得胶体二氧化硅作为起始材料完成‘一步变体’,其中二氧化硅与紫外光吸收化合物之间的反应在溶剂中进行,其中胶体二氧化硅悬浮于商购可得的产品(典型浓度为25-40wt%)。以这种方式,可以以不小于0.10mmol紫外光吸收化合物每克二氧化硅将紫外光吸收化合物加载到二氧化硅颗粒上。例如,已显示异丙醇是对反应有用的溶剂。乙二醇和异丙醇的混合物也是可行的。也可以使用其它溶剂,例如链烷醇和芳族烃如甲苯。二氧化硅的oh表面基团与紫外光吸收化合物(如2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮)的摩尔比为优选是8:1至2:1,例如3.5:1至2.5:1。允许反应在加热下(例如在回流下)进行约24小时。本发明也提供改善聚合物光稳定性的方法,其包括向聚合物中添加在其表面上具有紫外光吸收化合物的无机氧化物颗粒,如用上述紫外光吸收化合物官能化的二氧化硅和氧化锌颗粒。包含紫外光稳定剂添加剂的热塑性聚合物形成本发明的另一方面。可以将本发明的表面改性的无机颗粒加入到众多聚合物中,包括聚丙烯(均聚物和共聚物),聚乙烯例如高密度聚乙烯(hdpe)、中密度聚乙烯(mdpe)、低密度聚乙烯(ldpe)和线性低密度聚乙烯(lldpe),聚对苯二甲酸乙二醇酯(pet),和pdms。本发明添加剂的浓度不大于15wt%,例如1.0wt%至12.0wt%,优选1.0wt%至8.0wt%,基于聚合物制剂的重量。通过常规方法将添加剂引入到聚合物中以完成混配,例如通过使用挤出机如双螺杆挤出机进行。然后使用常规的膜/片材挤出(例如吹塑挤出和流延挤出)将所形成的粒料加工并成型为所需的制品如膜。制备引入添加剂的聚丙烯和lldpe膜的说明性步骤如下所示。通过如下测试引入添加剂的聚合物以测量紫外光吸收化合物到表面的迁移率:用清洁己烷定期洗涤和漂洗膜表面,和测量洗涤溶液的uv透射率以检测痕量的uv吸收材料和测量膜的uv透射率(保持在如下所述的不同条件下),以记录膜的紫外光吸收性能的任何劣化。以下报告的结果表明,紫外光吸收化合物即使在长期的储存期后也未从膜中迁移出。实施例方法:热重分析(tga):使用mettler-toledo仪器型号tga/sdta851进行tga分析。在氮气氛中将10mg样品在标准70μltga氧化铝坩埚中以10℃/min的加热速率从室温加热至约1000℃。二氧化硅表面上的uva分子的浓度根据以下等式计算:其中dc1是二氧化硅-间隔基在150-1000℃的分解含量(%),dc2是二氧化硅-间隔基-uva的分解含量,mw是uva的分子量(例如当为2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚时为265.31g/mol)。傅里叶变换红外光谱(ftir):使用的ftir仪器是scientific,nicoletis10。核磁共振(nmr)光谱:在brukeravanceii仪器,500mhz,cp-mas4mm上获取nmr光谱。x射线光电子能谱(xps):使用的x射线光电子能谱仪来自kratosanalyticalltd.(kratosaxisultra)。元素分析:使用的元素分析仪是perkinelmer的2400系列ii分析仪。下面的实施例组(1-3)说明了借助于具有四个(或六个)sih官能团的小的间隔基分子,紫外光吸收化合物(苯并三唑族)在二氧化硅颗粒表面上的共价结合,如下面描述的合成途径所示。制剂1制备二氧化硅-乙烯基制剂1a将10克二氧化硅(cab-o-silm5,cabotcorporation)和40克乙烯基三甲氧基硅烷(vtms;来自sigma-aldrich,目录号440221-97%)在装配有回流冷凝器和cacl2阱(trap)的三颈烧瓶中混合。使用油浴(150℃)将混合物加热至vtms的沸点(沸点123℃),并将其在该温度用机械搅拌器(225rpm)搅拌11小时。冷却至室温后,在具有烧结玻璃的过滤漏斗(p4)中用己烷(3×250ml)洗涤二氧化硅,将其在烘箱中在陪替氏培养皿中在165℃干燥半小时,得到9.56克二氧化硅-乙烯基(1,方案1)。制剂1b将二氧化硅(cab-o-silm5,cabotcorporation)在0.5吨压力下压制到体积减少约1.5。然后将10克二氧化硅和40克乙烯基三甲氧基硅烷(来自sigma-aldrich,目录号440221-97%)在装配有回流冷凝器和cacl2阱的250ml三颈烧瓶中混合。使用油浴(150℃)将混合物加热至vtms的沸点(沸点123℃),将其在该温度用机械搅拌器(278rpm)搅拌11小时。冷却至室温后,在具有烧结玻璃的过滤漏斗(p4)上用己烷(4×250ml)洗涤二氧化硅,将其在165℃在烘箱中干燥半小时,收集二氧化硅-乙烯基(1,方案1)。制剂1c:将二氧化硅(cab-o-silm5,cabotcorporation)在1吨压力下压制10分钟。然后将100克压制的二氧化硅和400克乙烯基三甲氧基硅烷(来自sigma-aldrich,目录号440221-97%)在装配有回流冷凝器和机械搅拌器(半月形,90mmptfe叶片)的1l圆柱形反应器中混合。使用加热套将混合物加热至vtms的沸点(沸点123℃),将其用机械搅拌器(300rpm)在该温度在氮气下搅拌19小时。冷却至室温后,向反应混合物中加入约400ml己烷,混合后,将所得悬浮液在1l具有烧结玻璃的过滤漏斗(p4)上过滤。用另外40ml己烷洗涤反应器,也过滤悬浮液。将残留在过滤器上的二氧化硅用另外己烷(3×800ml)洗涤,将其在160℃在烘箱中使用化学烧杯(14cm直径和7.5cm高)干燥3小时,收集二氧化硅-乙烯基(1,方案1)。通过nmr、ftir和xps分析表征二氧化硅-乙烯基(1,方案1)。nmr光谱如图1所示;在134ppm和128ppm处的两个峰归属于加入到二氧化硅表面的乙烯基的碳原子。在约48ppm处的峰归属于未反应的甲氧基(由于空间的考虑因素,在乙烯基三甲基硅烷的三个-och3基团中平均约两个-och3基团参与与二氧化硅表面的键形成反应)。图2显示了原始未改性的商业二氧化硅的ftir光谱(下面)和二氧化硅-乙烯基的ftir光谱(上面)。未改性的二氧化硅的特征峰是:在约3433cm-处的大的带,其归因于存在硅烷醇基团的o-h伸缩频率以及吸附的水;在1631cm-1处的尖锐带,其归属于分子水的弯曲振动;在1254-1026cm-1处的宽的强峰,其归属于si-o-si不对称键的伸缩振动。该峰在约980cm-1处具有一些“肩部”,其对应于si-oh的不对称振动。在806cm-1处的带,其归属于网络si-o-si对称键的伸缩振动;和在460cm-1处的带,其与网络si-o-si键的弯曲振动有关。乙烯基改性的二氧化硅的光谱在3060cm-1和3020cm-1处出现新的弱峰,其归因于si-ch=ch2基团的c-h带。在1600cm-1处的峰是典型的c=c振动,出现在1410cm-1处的峰归属于=ch2基团的c-h变形振动。在2986cm-1、2958cm-1和2987cm-1处的峰归属于残留甲氧基的c-h带(特征si-och3带,预期在约1000-1100cm-1处发现,其由强的si-o-si吸收掩蔽)。最后,应该指出的是,在改性的二氧化硅中,与原始样品相比,在3433cm-1处的宽的带显著减少。这种减少与在约980cm-1处的“肩部”消失一起明确表明,表面硅烷醇基团参与化学反应,即,乙烯基三甲氧基硅烷通过表面硅烷醇基团与二氧化硅表面共价连接。乙烯基三甲氧基硅烷成功连接至二氧化硅颗粒表面也通过下表的xps分析得到证实。预期二氧化硅表面的改性导致在二氧化硅表面上测量的氧/碳和硅/碳的比率降低。这确实通过下表1中列出的结果得以显示:表1实施例1制备二氧化硅-间隔基a4-uva[uva分子是2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基a4然后将10.00克制剂1a的二氧化硅-乙烯基(其在160℃干燥1h)、4.70克四(二甲基甲硅烷氧基)硅烷(a4;来自abcr,目录号ab111396-97%)和54克甲苯在500ml单颈烧瓶中用机械搅拌器混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(425μl)形式加入。将所得悬浮液在80℃用机械搅拌器(300rpm)搅拌5小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(2×500ml)和己烷(3×500ml)洗涤。将产物置于陪替氏培养皿中,在烘箱中在70℃烘箱干燥1小时,得到9.95克二氧化硅-a4(2,方案1)。制备二氧化硅-间隔基a4-uva将9.45克二氧化硅-间隔基a4、5.33克2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚{uva;来自sigma-aldrich,目录号559857-99%}和60克甲苯在250ml单颈烧瓶中用机械搅拌器混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(445μl)形式加入。将所得悬浮液在80℃搅拌16小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(3×400ml)和己烷(4×500ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到9.29克二氧化硅-间隔基a4-uva(3,方案1)。通过ftir、tga和元素分析表征如此形成的添加剂。图3中所示的ftir光谱在745cm-1处显示出峰,其归属于已成功连接至颗粒的uva分子。在2135cm-1和910cm-1处的峰是由于存在未与uva分子形成化学键的未反应的sih基团。根据上述方法,在tga分析的基础上计算施加到二氧化硅颗粒上的uva分子的浓度,得到其为0.26mmol/g(测量的dc1和dc2值分别为4.87%和11.40%)。由tga确定的浓度显示出与通过元素分析获得的结果相当良好的一致性[测量的%c:9.62;计算的浓度:0.28mmol/g(ce.a.)]实施例2制备二氧化硅-间隔基a6-uva[uva化合物是2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基a6然后将1克制剂1a的二氧化硅-乙烯基、1.32克聚(二甲基硅氧烷-共聚-甲基氢硅氧烷)、封端的三甲基甲硅烷基{a6;来自sigma-aldrich,目录号482196-平均mn为约950}和5.22克甲苯在40ml小瓶中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(50μl)形式加入。将所得悬浮液在80℃搅拌5小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(3×50ml)和己烷(4×50ml)洗涤。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到0.982克二氧化硅-间隔基a6。制备二氧化硅-间隔基a6-uva将750.8mg二氧化硅-间隔基a6、424.8mg2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚{uva;来自sigma-aldrich,目录号559857-99%}和4.78克甲苯在20ml小瓶中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(36μl)形式加入。将所得悬浮液在80℃搅拌8小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(4×40ml)和己烷(3×40ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到0.625克二氧化硅-间隔基a6-uva。通过ftir和tga表征如此形成的添加剂。图4中所示的ftir光谱在745cm-1处显示出特征峰,这表明在二氧化硅表面上存在uva分子。根据上述方法,在tga分析的基础上计算加载到二氧化硅颗粒上的uva分子的浓度,得到其为0.31mmol/g(测量的dc1和dc2值分别为6.08%和13.73%)。实施例3制备二氧化硅-间隔基a6y–uva[uva分子2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基a6y将1克制剂1a的二氧化硅-乙烯基、780mga6y(根据us5,691,435中描述的步骤制备)和3.6克甲苯在40ml小瓶中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(36μl)形式加入。将所得悬浮液在80℃搅拌5小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(4×40ml)和己烷(4×40ml)洗涤。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到0.984克二氧化硅-间隔基a6y。制备二氧化硅-间隔基a6y-uva将627.8mg二氧化硅-间隔基a6y、354.8mg2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚{uva;来自sigma-aldrich,目录号559857-99%}和2.26克甲苯在20ml小瓶中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(28μl)形式加入。将所得悬浮液在80℃搅拌8小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(4×40ml)和己烷(3×40ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到0.518克二氧化硅-间隔基a6y-uva。通过ftir和tga表征如此形成的添加剂。图5中所示的ftir光谱在745cm-1处显示出特征峰,这表明在二氧化硅表面上存在uva分子。根据上述方法,在tga分析的基础上计算存在于二氧化硅颗粒上的uva分子的浓度,得到其为0.51mmol/g(测量的dc1和dc2值分别为6.13%和19.04%)。下面的实施例组(4-8)说明了紫外光吸收化合物(苯并三唑族)借助于如下的长的间隔基共价结合到二氧化硅颗粒表面上:聚(甲基氢硅氧烷),其由约75个重复单元(即si-h官能团)组成。实施例4制备二氧化硅–间隔基mh30-uva[uva化合物是2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基mh30将制剂1a的二氧化硅-乙烯基(1克)在160℃干燥1h。将二氧化硅-乙烯基、3.29克andisilmh30(来自abspecialtysilicones)和2.6克甲苯在20ml烧瓶中用磁力搅拌器混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(45μl)形式加入。将所得悬浮液在50℃用磁力搅拌器搅拌8小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(50ml)和己烷(4×50ml)洗涤。将产物置于陪替氏培养皿中,在烘箱中在45℃干燥1小时,得到1.2克二氧化硅-mh30。制备二氧化硅-间隔基mh30-uva将0.5克二氧化硅-间隔基mh30、1.5克2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚(sigma-aldrich,目录号559857-99%)和3克甲苯在20ml烧瓶中用磁力搅拌器混合,然后加入pt-催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(80μl)形式加入。将所得悬浮液在80℃搅拌8小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(3×100ml)和己烷(3×100ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到0.45克二氧化硅-间隔基mh30-uva。通过ftir和tga表征如此形成的添加剂。ftir光谱如图6所示;注意到在745cm-1、1217cm-1和1264cm-1处存在强烈的特征峰,其归属于uva分子。根据上述方法,在tga分析的基础上计算存在于二氧化硅颗粒上的uva分子的浓度,得到其为2.35mmol/g(测量的dc1和dc2值分别为5.19%和41.63%)。实施例5制备二氧化硅-间隔基mh30-uva[uva分子是2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基mh30将50克制剂1c的二氧化硅-乙烯基(在160℃干燥1h)、106.4克andisilmh30和124.7克甲苯在装配有机械搅拌器(半月形,90mmptfe叶片)的1l圆柱形反应器中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(1.9ml)形式加入。将所得悬浮液在50℃用机械搅拌器(340rpm)搅拌16小时。冷却至室温后,加入300ml甲苯,剧烈混合反应混合物。然后使悬浮液静置半小时,沉降后,通过滗析溶剂分离固体。将产物用己烷(240ml,离心-滗析,3000rpm,每次10分钟)洗涤两次。最终滗析后,将固体引入至1l具有烧结玻璃的过滤漏斗(p4)中,将其用己烷(4×500ml)洗涤。蒸发最后一部分己烷,称重残留物,以确保洗掉所有未反应的聚合物。将产物置于陪替氏培养皿中,将其在烘箱中在50℃干燥1小时,得到45克二氧化硅-mh30。制备二氧化硅-间隔基mh30-uva将43.3克二氧化硅-间隔基mh30、129克2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚(由等量的购自sigma-aldrich的目录号559857-99%的材料和从如下所述的先前批料中回收的再生材料组成)和258克甲苯在装配有机械搅拌器(半月形,90mmptfe叶片)的1l圆柱形反应器中混合,然后加入pt-催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849}。催化剂以2%的甲苯溶液(7ml)形式加入。将所得悬浮液在80℃用机械搅拌器(340rpm)搅拌16小时。冷却至室温后,将产物在具有烧结玻璃的1l过滤漏斗(p4)上用甲苯(5×800ml)、己烷(3×800ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。合并滤液,蒸发浓缩,收集和重新使用未反应的2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1.5小时,得到55克二氧化硅-间隔基mh30-uva。实施例6制备二氧化硅-间隔基mh30-uva[uva分子是2-(2’-羟基苯基)苯并三唑衍生物]制备二氧化硅-间隔基mh30将14.39克制剂1c的二氧化硅-乙烯基(在160℃干燥1h)、31.58克andisilmh30和36.75克甲苯在装配有机械搅拌器的500ml三颈烧瓶中混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849;催化剂以2%的甲苯溶液(0.55ml)形式加入}。将所得悬浮液在50℃用机械搅拌器(340rpm)搅拌16小时。冷却至室温后,加入90ml甲苯,充分混合反应混合物。然后使悬浮液静置半小时,沉降后,通过滗析溶剂分离固体。将二氧化硅用己烷(4×250ml,离心-滗析,3000rpm,每次10分钟)洗涤四次。蒸发最后一部分己烷,称重残余物,以确保洗掉所有未反应的andisilmh30。将产物置于陪替氏培养皿中,在烘箱中在50℃干燥,得到14.82克二氧化硅-间隔基mh30。制备二氧化硅-间隔基mh30-uva将0.787克二氧化硅-间隔基mh30、0.787克2-(2h-苯并三唑-2-基)-4-甲基-6-(2-丙烯基)苯酚(sigma-aldrich,目录号559857-99%)和4.92克甲苯在20ml烧瓶中用机械搅拌器混合,然后加入pt催化剂{顺式-二氯二(二乙基硫醚)铂(ii),来自sigma-aldrich,目录号432849;催化剂以2%的甲苯溶液(105μl)形式加入}。将所得悬浮液在80℃搅拌16小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(2×50ml)、己烷(3×50ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有未反应的苯并三唑。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时。实施例7研究聚(甲基氢硅氧烷)作为间隔基用于将紫外光吸收化合物连接到二氧化硅颗粒表面上的性质在该实施例中报告的一组测量中,使用聚(甲基氢硅氧烷)(mh30,购自andisil)作为间隔基。2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑用作紫外光吸收化合物。光学性质:图7显示2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑、以及mh30与2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑之间反应的产物在紫外光-可见光范围内的透射光谱:加成反应在甲苯中在50℃在前述实施例中详述的相同条件下进行,得到具有侧基2-(3-丙基-2-羟基-5-甲基苯基)-2h-苯并三唑的聚(甲基氢硅氧烷)。为了得到光谱,使用50ppm在甲苯中的2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑,和类似地使用50ppm在甲苯中的反应产物。图7所示的光谱表明,2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑及其与mh30的反应产物的吸光度是相当的(在图7的光谱中分别表示tinb和mh30-tinb或mh30-tinb)。热性质:热稳定性使用tga分析(随着样品温度的升高,其测量样品的重量损失)研究。使以下样品进行tga分析,结果显示于图8中的热谱图中:单独的聚(甲基氢硅氧烷)(在图8的热谱图中称为“mh30”);聚(甲基氢硅烷烷)与2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑的反应产物,如上所述(在图8的热谱图中称为“mh30-uva”);二氧化硅颗粒与mh30的反应产物,如先前实施例中所述(在图8的热谱图中称为“sil-mh30”);和如先前实施例中所述制备的本发明的添加剂,由具有连接至二氧化硅表面的mh30间隔基的二氧化硅颗粒和化学键合至间隔基的2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑组成(在图8的热谱图中称为“sil-mh30-uva”)。tga表明聚(甲基氢硅氧烷)(即mh30)的分解在约250℃的温度开始。mh30的热稳定性在mh30连接至二氧化硅颗粒表面时降低,这是因为“sil-mh30”缀合物的分解在150℃开始。因此,偶联至二氧化硅颗粒对mh30的热稳定性具有不利影响。然而,2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑与mh30链的化学键合对热稳定性具有有利影响,其中反应产物mh30-uva显示出增加的热稳定性,这是因为在约450℃的较高温度发生分解。最重要的是,在本发明的添加剂sil-mh30-uva中,2-(3-烯丙基-2-羟基-5-甲基苯基)-2h-苯并三唑对mh30的热稳定性的有利影响超过了偶联至二氧化硅颗粒的不利影响。实施例8制备二氧化硅-uva[uva是2-羟基二苯甲酮衍生物]将1.00克二氧化硅(cab-o-silm5,cabotcorporation)、0.96克2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯甲酮(95%,来自gelest)和17.40克甲苯在装配有回流冷凝器和cacl2阱的50ml烧瓶中用磁力搅拌器混合。使用油浴(120℃)将混合物加热至甲苯的沸点(沸点111℃),将其用磁力搅拌器在该温度搅拌5小时。冷却至室温后,将产物在具有烧结玻璃的过滤漏斗(p4)上用甲苯(3×250ml)和己烷(2×250ml)洗涤。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有的羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮。将产物置于陪替氏培养皿中,在烘箱中在70℃干燥1小时,得到1.08克二氧化硅-uva。通过ftir和tga表征如此形成的添加剂。ftir光谱如图9所示;在1626.7cm-1处的峰对应于uva分子内的羰基的强烈带。得到基于tga分析计算的二氧化硅颗粒上存在的uva分子的浓度为0.315mmol/g。实施例9制备二氧化硅-uva[uva是2-羟基二苯甲酮衍生物]在该实施例中报道的实验中,使用在溶剂中的胶体二氧化硅(来自evonikindustriesag)。二氧化硅起始原料由单分散二氧化硅颗粒群在异丙醇中的悬浮液(30wt%-40wt%)组成。通过tga测量二氧化硅起始原料表面上的硅烷醇基团的浓度,得到其为1.81mmol/g(在室温至1100℃的温度范围内进行溶剂蒸发后进行tga)。二氧化硅颗粒与紫外光吸收化合物2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮的反应在异丙醇中在回流温度(82℃)发生。二氧化硅的oh表面基团与2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮的摩尔比为3:1,使得反应在回流下进行24小时。在反应物比率和反应时间的所选条件下,悬浮液通过反应保持其透明度,表明没有经历聚集。通过冷冻透射电镜(cryo-tem)分析(在异丙醇中浓度为5%)检查颗粒,图10中的冷冻透射电镜图像显示离散颗粒的可分散性。紫外光吸收化合物的浓度经测量(tga)为0.15mmol/g。实施例10制备zno-uva[uva是2-羟基二苯甲酮衍生物]将36.03克zno(20,来自umicorezincchemicals)、53.60克2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮和144.13克甲苯用机械搅拌器在装配有回流冷凝器和cacl2阱的250ml烧瓶中搅拌。使用油浴(120℃)将混合物加热至甲苯的沸点(沸点111℃),将其在该温度搅拌(250rpm)65小时。冷却至室温后,在带有烧结玻璃的过滤漏斗上用甲苯和己烷洗涤zno颗粒。测量最后一部分己烷的uv吸光度(250nm-450nm),以确保洗掉所有uva。将zno在70℃在烘箱中在陪替氏培养皿中干燥1小时,得到35.4克zno-uva。通过ftir、tga和xps分析表征如此形成的添加剂。ftir光谱如图11所示(下面的光谱对应于zno,上面的曲线对应于uva改性的zno)。可以看出,uva改性的zno的光谱在2967cm-1和2920cm-1处显示出新的弱峰,其归因于c-h带。在1628.5cm-1处的强峰归属于uva分子内的羰基。指纹区也表现出uva分子的新特征峰。约1118-1023cm-1的宽峰归因于si-o-si和si-o-c带;si-o-zn吸收带位于916cm-1处。tga分析表明,uva分子在颗粒表面上的经计算的浓度是0.09mmol/g。xps用于分析原始zno颗粒和表面改性的zno颗粒的组成,以确定颗粒表面上元素的原子百分比。数据分别列于表2和表3中:表2(原始zno)名称峰befwhmev面积(p)cps.ev原子%qc1s284.751.202971.989.341o1s529.801.1240861.8146.811zn2p31020.941.50193072.4043.851表3(zno-uva)名称峰befwhmev面积(p)cps.ev原子%qc1s284.921.782666.7425.811o1s530.932.2013462.4745.781zn2p31023.482.3439986.9624.651si2p101.981.41321.543.761uva分子2-羟基-4-(3-三乙氧基甲硅烷基丙氧基)-二苯基酮与zno颗粒通过以下所示反应的连接:由位于约102ev的新峰表示,其归属于硅的结合能(be)(注意uva分子含有si原子)。在原始zno颗粒中测量的氧和锌之间的原子比是约1:1;由于uva分子的氧原子和表面锌的掩蔽,该比率在反应产物中增至约2:1。反应产物中的碳含量高于商业zno颗粒测量的碳含量。最后,对于zn2p线的xps光谱的反卷积显示,在反应产物中存在锌的三种不同的结合能(约1022.1ev、约1023.3ev和约1024.3ev),如表4中所列。另一方面,原始zno颗粒的光谱仅包含一个结合能带(对应于zn的单一化学状态)。表4名称峰befwhmev面积(p)cps.ev原子%qzn2p扫描b1022.061.886524.6216.311zn2p扫描c1023.291.5414095.6335.271zn2p扫描a1024.261.6919336.7148.411实施例11聚丙烯膜制备该实施例说明了使用本发明的添加剂制备聚丙烯膜。膜的组成由95%聚丙烯和5%添加剂(按重量计)组成。聚丙烯均聚物(来自'reliancepolymers'的repolh034sg)和实施例1中制备的本发明的添加剂通过在eurolabdigital16('prism')双螺杆同向旋转挤出机(d=16mm,l/d=24)中使用200-225℃的均匀温度分布和以250rpm的螺杆速度操作熔融混合而混配,得到100克批料大小的混配物。将这些材料在塑料袋中混合,并且在相同的上游进料口加入到挤出机。在室温将熔融混配物线料在水下连续冷却以凝固,然后用造粒机将其切成粒料。将预混配的粒料在进一步加工之前干燥(70℃,过夜,真空)。然后使用200-220℃的温度分布将粒料进料至microtruder('randcastle')流延膜单螺杆挤出机(d=13mm,l/d=20)。该机器以70rpm的螺杆速度操作,得到壁厚50微米、宽度150mm的膜。通过如下测试引入添加剂的聚合物以测量紫外光吸收化合物到表面的迁移:用清洁己烷定期洗涤和漂洗膜表面,和测量洗涤溶液的uv透射率以检测痕量的uv吸收材料和测量膜的uv透射率,以记录膜的紫外光吸收性能的任何劣化。在制备膜后立即记录、以及在制备膜后约两年记录图12中所示的uv光谱。应注意,在己烷中第一次测量后,在下列条件下将膜储存在玻璃小瓶中(参见图12中的附图标记膜“1”、膜“2”和膜“3”):膜#1,在空气中@30℃膜#2,在95%乙醇中@30℃膜#3,在空气中@60℃结果表明,在所有情况下,uv阻挡水平不随时间推移而降低,表明借助于本发明的添加剂有效地抑制了紫外光吸收化合物的表面迁移。实施例12lldpe膜制备该实施例说明了通过将本发明的添加剂引入到7μm厚的芯层中制备取向的15μm厚的多层膜(三层)。膜的组成由93%lldpe和7%添加剂(按重量计)组成。lldpe(novachemicalsfp112-a)和实施例4中制备的本发明的添加剂如下进行混配:将其在16mm双螺杆挤出机(prism,england)(所述螺杆机l/d比为25:1,利用160-225℃的温度分布,并且以200-230rpm的螺杆速度操作)中熔融混合,得到1500克批料大小的混配物。将材料混合并通过相同的上游进料口进料至挤出机。在室温将熔融混配物线料在水下连续冷却以凝固,然后用造粒机将其切成粒料。将预混配的粒料在进一步加工之前干燥。将预混配的材料样品进料至microtruder('randcastle')流延膜单螺杆挤出机(d=13mm,l/d=20)中以制备三层流延膜。对于挤出机a,该过程在200-220℃的温度分布进行,对于挤出机c,该过程在200-230℃的温度分布进行。对于挤出机a,机器以64rpm的螺杆速度操作,对于挤出机c,机器以115rpm的螺杆速度操作,得到总厚度为400μm和宽度为150mm的三层膜。然后将400μm膜拉伸至最终尺寸。在制备膜后的几天记录uv光谱,然后在约十三个月之后再次记录uv光谱(膜“1”、膜“2”和膜“3”具有与上述相同的含义)。如图13所示的uv光谱表明,在这约一年的时间段内,没有发生添加剂到膜表面的迁移,这表明在膜制备后立即由膜显示出的紫外光阻挡与在制备膜一年之后由膜显示出的紫外光阻挡基本相当。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1