环状DNA的扩增方法与流程
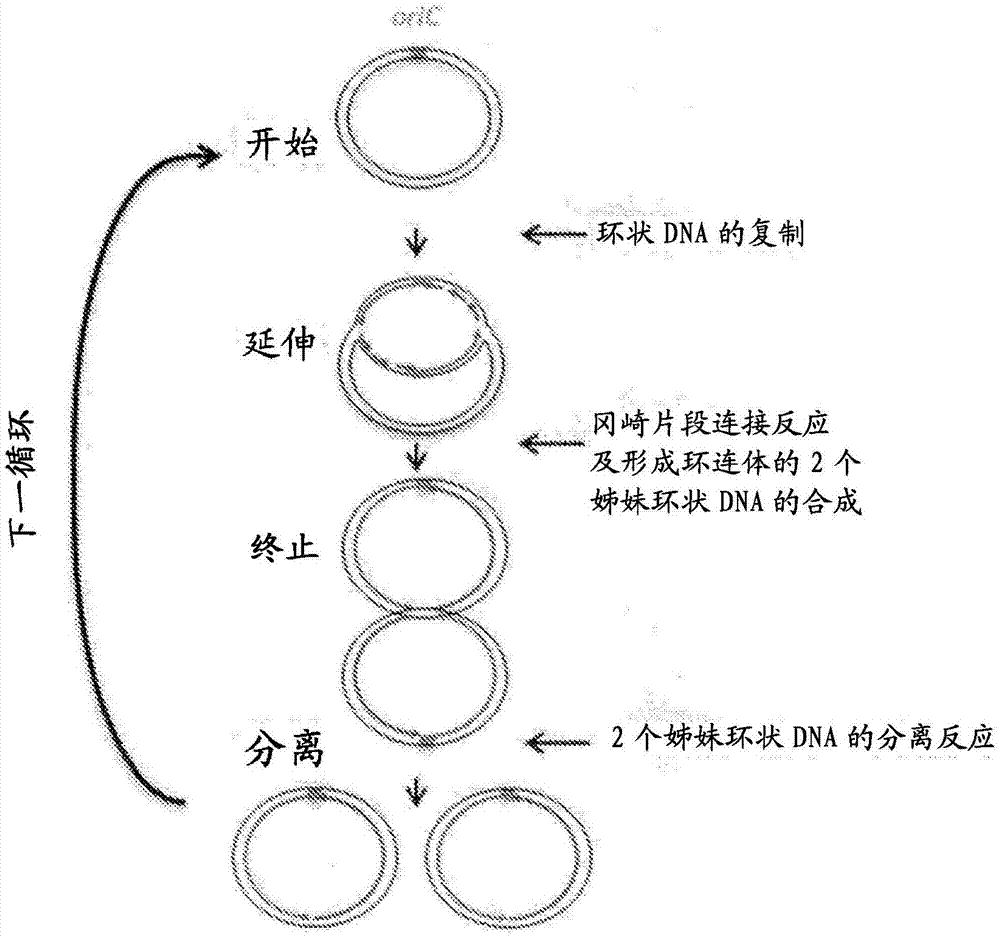
本发明涉及环状dna的扩增方法。更具体而言,涉及可在无细胞系统中指数扩增环状dna的方法。
背景技术:
:成为生物技术发展的基础的dna克隆技术是使由dna片段的切割粘贴调制的环状dna在大肠杆菌等的细胞内作为质粒扩增的方法。在利用使用细胞的dna克隆技术而扩增环状dna时,细胞培养及扩增产物的提取-纯化等的烦琐的顺序变得必要。另外,由于有为了进行使用细胞的dna克隆而制出基因重组生物的必要,可实验的环境上有限制。作为在试管内扩增dna的方法,一般使用聚合酶链式反应(pcr)。但是,由利用pcr的试管内dna扩增法无法直接扩增环状dna。作为环状dna的试管内扩增法,有滚环扩增法(rca)等(非专利文献1、专利文献1、专利文献2、专利文献3)。但是,为了用滚环扩增法扩增环状dna,有每次设计对目标dna具有特异性的引物的必要。另外,由滚环扩增法的直接的扩增产物是直链型dna,为了使得到的扩增产物环化,与重组酶温育等的进一步的环化工序变得必要。也报告了在复制大肠杆菌的小染色体(oric环状dna)之后,将其分离,得到单体的环状复制产物的方法(非专利文献2~5)。但是,在这些文献中使用的反应条件中,实验显示作为环状dna分子的复制效率止于所加的模板dna的15-40%左右,作为扩增量未达倍增(非专利文献3~6)。再者,在这些文献中作为模板使用的环状dna的尺寸止于不足10kbp。这样,为了用以往的试管内dna扩增法扩增环状dna,引物向模板dna的结合是必要的,扩增产物是直链型dna,另外,能扩增的dna尺寸止于数kbp。再者,在要使用大肠杆菌小染色体复制系统来产生环状的扩增产物时,有模板环状dna不扩增翻倍的问题。【现有技术文献】【专利文献】专利文献1:特开2005-229950专利文献2:特开2008-161182专利文献3:特表2012-501173【非专利文献】非专利文献1:fakruddinmetal.、jpharmbioalliedsci.2013、5:245-252非专利文献2:pengh&marianskj.pnas.1993、90:8571-8575非专利文献3:hiasah&marianskj.jbiolchem.1994、269:32655-32659非专利文献4:funnellbetal.、jbiolchem.1986、261:5616-5624非专利文献5:hiasahetal.、jbiolchem.1994、269:2093-2099非专利文献6:hiasah&marianskj.jbiolchem.1994、269:26959-26968【发明的概要】【发明要解决的课题】本发明旨在提供可在无细胞系统中简便并且指数扩增环状dna、特别是长链环状dna的方法。【用于解决课题的手段】本发明人等为解决上述课题而重复锐意研究的结果发现,通过使有复制起始序列(originofchromosome(oric))的环状dna与含以下物质的反应液混合而生成的反应混合物反应:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;“复制的开始(dna双键断裂)-延伸(复制叉进行)-复制的姊妹dna的分离(decatenation)”的循环重复,可指数扩增环状dna。即,虽不限于此,但本发明包含以下的实施方式的发明。[1]环状dna的扩增方法,其包括以下的工序:(1)形成成为模板的环状dna和含以下物质的反应液的反应混合物的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric));及(2)在等温条件下保温在工序(1)中形成的反应混合物的工序。[2]环状dna的扩增方法,其包括以下的工序:(1)形成成为模板的环状dna和含以下物质的反应液的反应混合物的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric));及(2)将在工序(1)中形成的反应混合物在重复30℃以上的温育及27℃以下的温育的温度循环下温育的工序。[3]上述[1]或[2]所述的方法,其中反应液还含蛋白质的非特异吸附抑制剂、及/或核酸的非特异吸附抑制剂。[4]上述[1]或[2]所述的方法,其中反应液还含直链状dna特异性外切核酸酶及/或recg型解螺旋酶。[5]上述[1]或[2]所述的方法,其中反应液还含铵盐。[6]上述[1]或[2]所述的方法,其中第一酶组含具有dnaa活性的酶、1种以上的核样体蛋白质、有dna促旋酶活性的酶或酶组、单链dna结合蛋白质(single-strandbindingprotein(ssb))、有dnab型解螺旋酶活性的酶、有dna解螺旋酶装载体活性的酶、有dna引发酶活性的酶、有dna夹活性的酶、及有dna聚合酶iii*活性的酶或酶组的组合,第二酶组含具有dna聚合酶i活性的酶及有dna连接酶活性的酶的组合,第三酶组含具有拓扑异构酶iii活性的酶及/或有拓扑异构酶iv活性的酶。[7]上述[6]所述的方法,其中第二酶组还含具有rna酶h活性的酶。[8]上述[6]所述的方法,其中第三酶组还含具有recq型解螺旋酶活性的酶。[9]上述[6]所述的方法,其中在第一酶组中,1种以上的核样体蛋白质是ihf或hu,有dna促旋酶活性的酶或酶组是由gyra及gyrb构成的复合物,有dnab型解螺旋酶活性的酶是dnab解螺旋酶,有dna解螺旋酶装载体活性的酶是dnac解螺旋酶装载体,有dna引发酶活性的酶是dnag引发酶,有dna夹活性的酶是dnan夹,有dna聚合酶iii*活性的酶或酶组是含dnax、hola、holb、holc、hold、dnae、dnaq、及hole之任一者的酶或酶组。[10]上述[1]所述的方法,其中在工序(2)中的等温条件是含在25℃~50℃的范围内的一定的温度。[11]上述[1]或[2]所述的方法,其中反应液还含recg型解螺旋酶及/或单链dna特异性外切核酸酶。[12]上述[1]或[2]所述的方法,其中反应液还含直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶。[13]上述[1]或[2]所述的方法,其中反应液还含dna的稳定化因子。[14]上述[1]或[2]所述的方法,其中工序(1)含下列工序:(1-1)对含以下物质的反应液进行预温育的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;及(1-2)形成所述反应液和成为模板的环状dna的反应混合物的工序。[15]上述[1]或[2]所述的方法,其中在油包水滴型乳液内进行工序(2)。[16]上述[1]或[2]所述的方法,其继工序(2)而还含(3)进行反应后处理的工序,其中所述反应后处理是:(i)用不含第一至第三酶组的反应液稀释为五倍以上之后,再保温的处理;(ii)由直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶的处理;及/或(iii)由缺口修复酶的处理。[17]环状dna的扩增用组合物,其含:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源。[18]上述[17]所述的组合物,其中还含蛋白质的非特异吸附抑制剂、及/或核酸的非特异吸附抑制剂。[19]上述[17]所述的组合物,其中还含直链状dna特异性外切核酸酶及/或recg型解螺旋酶。[20]上述[17]所述的组合物,其中还含recg型解螺旋酶及/或单链dna特异性外切核酸酶。[21]上述[17]所述的组合物,其中还含直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶。[22]上述[17]所述的组合物,其中还含dna的稳定化因子。[23]环状dna的扩增用试剂盒,其含下列物质的组合:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源。[24]上述[23]所述的试剂盒,其中还含蛋白质的非特异吸附抑制剂、及/或核酸的非特异吸附抑制剂的组合。[25]上述[23]所述的试剂盒,其中还含直链状dna特异性外切核酸酶及/或recg型解螺旋酶的组合。[26]上述[23]所述的试剂盒,其中还含recg型解螺旋酶及/或单链dna特异性外切核酸酶。[27]上述[23]所述的试剂盒,其中还含直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶。[28]上述[23]所述的试剂盒,其中还含dna的稳定化因子。[29]上述[23]所述的试剂盒,其中还含缺口修复酶。[30]环状dna的扩增方法,其包括形成含以下物质的反应液和成为模板的环状dna的反应混合物的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源,其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric))的,重复复制循环,指数扩增环状dna。[31]环状dna的扩增方法,其包括以下的工序:(1)形成成为模板的环状dna和含以下物质的反应液的反应混合物的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric));及(2)将工序(1)中形成的反应混合物在指定的温度范围保温的工序。[32]环状dna的扩增用试剂盒,其含下列物质的组合:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源,以及记载了用于实施通过在含上述的组合的反应液和成为模板的环状dna的反应混合物中重复复制循环来指数扩增环状dna的方法的指示的说明书。[33]上述[1]、[2]、[30]及[31]之任一项所述的方法,其中反应液中还含trna。[34]上述[17]所述的组合物,其中反应液中还含trna。[35]上述[23]或[32]所述的试剂盒,其中反应液中还含trna。[36]上述[1]、[2]、[30]及[31]之任一项所述的方法,其中反应液中还含100mm以上的碱金属离子源。[37]上述[17]所述的组合物,其中反应液中还含100mm以上的碱金属离子源。[38]上述[23]或[32]所述的试剂盒,其中反应液中还含100mm以上的碱金属离子源。[39]上述[1]、[2]、[30]及[31]之任一项所述的方法,其中环状dna扩增至少10倍。【发明的效果】由本发明提供不使用大肠杆菌细胞或质粒载体而可简便并且指数扩增环状dna、特别是长链环状dna的方法。根据本发明,扩增环状dna不需要引物,超200kb的长链环状dna的扩增也是可能的。进而,根据本发明的方法,模板环状dna从微少到1分子也能扩增环状dna。另外,由本发明得到的扩增产物是与原来的模板相同的环状结构原样的拷贝。再者,也可在连接多个dna片段之后,如果直接加到所述反应系统中,则仅特异性扩增由连接环化的dna而调制。【附图的简单的说明】【图1】图1显示根据本发明的复制循环的模型。【图2】图2显示由使用gibson组装法的试管内连接环化的dna的结构。【图3】图3显示对以9.6kb的环状dna作为模板使用时的每反应时间的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。【图4】图4显示对以200kb及80kb的长链环状dna作为模板使用时的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。图4a显示以200kb的长链环状dna(15pm、20ng)作为模板使用时的,每反应时间的扩增产物的结果。图4b显示以80kb(15pm、8ng)及200kb(5pm、6.7ng)的长链环状dna作为模板使用时的反应3小时后的扩增产物的结果。【图5】图5显示对以由使用gibson组装法的试管内连接环化的dna作为模板使用时的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。【图6】图6显示以微量(1分子水平)的9.6kb的环状dna作为模板使用的扩增实验的结果。图6a显示对以9.6kb的环状dna作为模板使用时的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。图6b是显示将扩增产物的dna量由picogreen法或大肠杆菌转化法定量,显示其扩增程度的结果的坐标图。【图7】图7是显示相对于以9.6kb的环状dna作为模板使用时的扩增时间的扩增的环状dna分子数的坐标图。【图8】图8是显示自混合物的单一的环状dna克隆的扩增试验结果的图。图8a是对于环状dna的混合物的稀释的模式图。图8b显示对稀释环状dna的混合物而扩增时的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。【图9】图9是显示环状dna的传代扩增试验结果的图。图9a是关于实验顺序的模式图。图9b显示将扩增反应后的dna产物在新的反应液中稀释而重复10次导致再次扩增的传代扩增时的结果。扩增产物由琼脂糖电泳及sybrgreen检测。【图10】图10显示对以80kb的环状dna作为模板使用、添加recg及直链状dna特异性外切核酸酶时的扩增产物进行琼脂糖电泳,由sybrgreen检测的结果。【图11】图11是显示实施例7的条件a的结果的坐标图。【图12】图12是显示实施例7的条件b的结果的坐标图。【图13】图13是显示实施例7的条件c(gtp、ctp及utp量的探讨)的结果的坐标图、及凝胶电泳的照片。【图14】图14是显示实施例7的条件d(ihf量的探讨)的结果的坐标图。【图15】图15是显示实施例7的条件e(topoiv量的探讨)的结果的坐标图。【图16】图16是显示实施例7的条件f(dna促旋酶量的探讨)的结果的坐标图。【图17】图17是显示实施例7的条件g(dna聚合酶iii*量的探讨)的结果的坐标图。【图18】图18是显示实施例7的条件h(碱金属离子源量的探讨)的结果的坐标图。【图19】图19是显示实施例7的条件i(蛋白质的非特异吸附抑制剂及/或核酸的非特异吸附抑制剂的量的探讨)的结果的坐标图。【图20】图20是显示实施例7的条件j(有dnaa活性的酶的量的探讨)的结果的坐标图。【图21】图21是显示实施例7的条件k(有dna连接酶活性的酶的量的探讨)的结果的坐标图。【图22】图22是显示实施例7的条件l(ssb量的探讨)的结果的坐标图、及凝胶电泳的照片。【图23】图23是显示实施例7的条件m(有dna聚合酶i活性的酶的量的探讨)的结果的坐标图。【图24】图24是显示实施例7的条件n(有dnab型解螺旋酶活性的酶及有dna解螺旋酶装载体活性的酶的量的探讨)的结果的坐标图。【图25】图25是显示实施例7的条件o(有rna酶h活性的酶的量的探讨)的结果的坐标图。【图26】图26是显示实施例7的条件p的结果的坐标图。【图27】图27是显示实施例7的条件q(第三酶组的酶的组成及量的探讨)的结果的,凝胶电泳的照片及坐标图。【图28】图28是显示实施例7的条件r的结果的坐标图。【图29】图29是显示实施例7的条件s的结果的坐标图。【图30】图30是显示探讨碱金属离子源的添加的效果的结果的凝胶电泳的照片。【图31】图31是显示探讨由预温育的扩增反应的效率化的结果的凝胶电泳的照片。【图32】图32是显示检测添加recg及recj时的扩增产物的结果的,凝胶电泳的照片。【图33】图33是显示检测添加recbcd及exoi时的扩增产物的结果的凝胶电泳的照片。【图34】图34是显示扩增反应后,检测用recbcd及exoi处理时的扩增产物的结果的凝胶电泳的照片。【图35】图35是显示扩增反应后,检测用缺口修复酶处理时的扩增产物的结果的凝胶电泳的照片。【图36】图36是显示探讨使用长链环状dna的稳定化因子时的扩增反应的效率的结果的凝胶电泳的照片。【图37】图37是显示探讨在油包水滴型乳液内的环状dna的扩增反应的结果的凝胶电泳的照片。【图38】图38是显示检测在伴随温度循环的环状dna的扩增反应中的扩增产物的结果的凝胶电泳的照片。【具体实施方式】接下来,对本发明具体进行说明,但本发明不限于这些。本说明书中只要是不特别定义,本发明中关联使用的科学用语及技术用语就应有本领域技术人员一般理解的含意。<环状dna>作为模板使用的环状dna优选为双键。作为模板只要使用的环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric)),就不特别限制,可例示向微生物的环状染色体等的天然的环状dna、将天然的环状dna由酶处理等切断的等连接别的dna片段,使其环化的环状dna、全部人工合成的环状dna等。作为能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric))(以下,有时简称为“复制起始序列”),可从ncbi(http://www.ncbi.nlm.nih.gov/)等的公共数据库得到存在于例如大肠杆菌、枯草芽孢杆菌等的细菌的公知的复制起始序列。另外,通过克隆能与有dnaa活性的酶结合的dna片段,对其碱基序列进行解析而也可得到复制起始序列。在本发明中作为模板使用的环状dna可为原本含复制起始序列的环状dna,也可向原本不含复制起始序列的环状dna导入复制起始序列。在本发明中作为模板使用的环状dna可对应于目的而含卡那霉素、氨苄西林、四环素等的药剂抗性标志物基因序列。在本发明中作为模板使用的环状dna可被纯化,也可为含环状dna的菌体提取物等的悬浮液的形态。另外,可以1种环状dna作为模板使用,也可以如例如dna库一样的多种环状dna的混合物在1个试管内作为模板使用。不限于本发明中作为模板使用的环状dna的长度,可设为例如1kb(1000碱基长)以上、5kb(5000碱基长)以上、8kb(8,000碱基长)以上、10kb(10,000碱基长)以上、50kb(50,000碱基长)以上、100kb(100,000碱基长)以上、200kb(200,000碱基长)以上、500kb(500,000碱基长)以上、1000kb(1,000,000碱基长)以上、或者2000kb(2,000,000碱基长)以上的长度。<第一、第二及第三酶组>1.第一酶组在本说明书中第一酶组是指催化环状dna的复制的酶组。作为催化环状dna的复制的第一酶组,可使用例如kagunijm&kornberga.cell.1984、38:183-90中记载的酶组。具体而言,作为第一酶组,可例示选自以下的酶或酶组中的一种以上:有dnaa活性的酶、1种以上的核样体蛋白质、有dna促旋酶活性的酶或酶组、单链dna结合蛋白质(single-strandbindingprotein(ssb))、有dnab型解螺旋酶活性的酶、有dna解螺旋酶装载体活性的酶、有dna引发酶活性的酶、有dna夹活性的酶、及有dna聚合酶iii*活性的酶或酶组、或者所述酶或酶组的全部的组合。作为有dnaa活性的酶,只要是有与作为大肠杆菌的起始物蛋白质的dnaa同样的起始物活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dnaa。大肠杆菌来源的dnaa作为单体而在反应液中可以1nm~10μm的范围含,也可优选为以1nm~~5μm、1nm~3μm、1nm~1.5μm、1nm~1.0μm、1nm~500nm、50nm~200nm、50nm~150nm的范围含,但不限于此。核样体蛋白质是指核样体中所含的蛋白质。在本发明中使用的1种以上的核样体蛋白质只要是有与大肠杆菌的核样体蛋白质同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的ihf、即ihfa及/或ihfb的复合物(异源二聚体或同源二聚体)、或大肠杆菌来源的hu、即hupa及hupb的复合物。大肠杆菌来源的ihf作为异源/同源2聚体而在反应液中可以5nm~400nm的范围含,也可优选为以5nm~200nm、5nm~100nm、5nm~50nm、10nm~50nm、10nm~40nm、10nm~30nm、的范围含,但不限于此。大肠杆菌来源的hu在反应液中可以1nm~50nm的范围含,也可优选为以5nm~50nm、5nm~25nm的范围含,但不限于此。作为有dna促旋酶活性的酶或酶组,只要是有与大肠杆菌的dna促旋酶同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如由大肠杆菌来源的gyra及gyrb构成的复合物。由大肠杆菌来源的gyra及gyrb构成的复合物作为异源4聚体而在在反应液中可以20nm~500nm的范围含,也可优选为以20nm~400nm、20nm~300nm、20nm~200nm、50nm~200nm、100nm~200nm的范围含,但不限于此。作为单链dna结合蛋白质(single-strandbindingprotein(ssb)),只要是有与大肠杆菌的单链dna结合蛋白质同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的ssb。大肠杆菌来源的ssb作为同源4聚体而在反应液中可以20nm~1000nm的范围含,也可优选为以20nm~500nm、20nm~300nm、20nm~200nm、50nm~500nm、50nm~400nm、50nm~300nm、50nm~200nm、50nm~150nm、100nm~500nm、100nm~400nm的范围含,但不限于此。作为有dnab型解螺旋酶活性的酶,只要是有与大肠杆菌的dnab同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dnab。大肠杆菌来源的dnab作为同源6聚体而在在反应液中可以5nm~200nm的范围含,也可优选为以5nm~100nm、5nm~50nm、5nm~30nm的范围含,但不限于此。作为有dna解螺旋酶装载体活性的酶,只要是有与大肠杆菌的dnac同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dnac。大肠杆菌来源的dnac作为同源6聚体而在在反应液中可以5nm~200nm的范围含,也可优选为以5nm~100nm、5nm~50nm、5nm~30nm的范围含,但不限于此。作为有dna引发酶活性的酶,只要是有与大肠杆菌的dnag同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dnag。大肠杆菌来源的dnag作为单体而在反应液中可以20nm~1000nm的范围含,也可优选为以20nm~800nm、50nm~800nm、100nm~800nm、200nm~800nm、250nm~800nm、250nm~500nm、300nm~500nm的范围含,但不限于此。作为有dna夹活性的酶,只要是有与大肠杆菌的dnan同样的活性的酶,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dnan。大肠杆菌来源的dnan作为同源2聚体而在在反应液中可以10nm~1000nm的范围含,也可优选为以10nm~800nm、10nm~500nm、20nm~500nm、20nm~200nm、30nm~200nm、30nm~100nm的范围含,但不限于此。作为有dna聚合酶iii*活性的酶或酶组,只要是有与大肠杆菌的dna聚合酶iii*复合物同样的活性的酶或酶组,其生物学来源就无特别限制,可适宜地使用例如含大肠杆菌来源的dnax、hola、holb、holc、hold、dnae、dnaq、及hole之任一者的酶组、优选为含大肠杆菌来源的dnax、hola、holb、及dnae的复合物的酶组、再优选为含大肠杆菌来源的dnax、hola、holb、holc、hold、dnae、dnaq、及hole的复合物的酶组。大肠杆菌来源的dna聚合酶iii*复合物作为异源多聚体而在反应液中可以2nm~50nm的范围含,也可优选为以2nm~40nm、2nm~30nm、2nm~20nm、5nm~40nm、5nm~30nm、5nm~20nm的范围含,但不限于此。2.第二酶组在本说明书中第二酶组是指催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的酶组。在本发明中,形成环连体的2个姊妹环状dna是指由dna复制反应合成的2个环状dna处于连接的状态。作为催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组,可例示选自例如有dna聚合酶i活性的酶、有dna连接酶活性的酶、及有rna酶h活性的酶的1种以上的酶或所述酶的组合。作为有dna聚合酶i活性的酶,只要是有与大肠杆菌的dna聚合酶i同样的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dna聚合酶i。大肠杆菌来源的dna聚合酶i作为单体而在反应液中可以10nm~200nm的范围含,也可优选为以20nm~200nm、20nm~150nm、20nm~100nm、40nm~150nm、40nm~100nm、40nm~80nm的范围含,但不限于此。作为有dna连接酶活性的酶,只要是有与大肠杆菌的dna连接酶同样的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的dna连接酶或t4噬菌体的dna连接酶。大肠杆菌来源的dna连接酶作为单体而在反应液中可以10nm~200nm的范围含,也可优选为以15nm~200nm、20nm~200nm、20nm~150nm、20nm~100nm、20nm~80nm的范围含,但不限于此。作为有rna酶h活性的酶,只要是有分解rna:dna杂交体的rna链的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的rna酶h。大肠杆菌来源的rna酶h作为单体而在反应液中可以0.2nm~200nm的范围含,也可优选为以0.2nm~200nm、0.2nm~100nm、0.2nm~50nm、1nm~200nm、1nm~100nm、1nm~50nm、10nm~50nm的范围含,但不限于此。3.第三酶组在本说明书中第三酶组是指催化2个姊妹环状dna的分离反应的酶组。作为催化2个姊妹环状dna的分离反应的第三酶组,可使用例如pengh&marianskj.pnas.1993、90:8571-8575中记载的酶组。具体而言,作为第三酶组,可例示选自以下的1种以上的酶或所述酶的组合:有拓扑异构酶iv活性的酶、有拓扑异构酶iii活性的酶、及有recq型解螺旋酶活性的酶。作为有拓扑异构酶iii活性的酶,只要是有与大肠杆菌的拓扑异构酶iii同样的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的拓扑异构酶iii。大肠杆菌来源的拓扑异构酶iii作为单体而在反应液中可以20nm~500nm的范围含,也可优选为以20nm~400nm、20nm~300nm、20nm~200nm、20nm~100nm、30~80nm的范围含,但不限于此。作为有recq型解螺旋酶活性的酶,只要是有与大肠杆菌的recq同样的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的recq。大肠杆菌来源的recq作为单体而在反应液中可以20nm~500nm的范围含,也可优选为以20nm~400nm、20nm~300nm、20nm~200nm、20nm~100nm、30~80nm的范围含,但不限于此。作为有拓扑异构酶iv活性的酶,只要是有与大肠杆菌的拓扑异构酶iv同样的活性,其生物学来源就无特别限制,可适宜地使用例如作为parc和pare的复合物的大肠杆菌来源的拓扑异构酶iv。大肠杆菌来源的拓扑异构酶iv作为异源4聚体而在反应液中可以0.1nm~50nmm的范围含,也可优选为以0.1nm~40nm、0.1nm~30nm、0.1nm~20nm、1nm~40nm、1nm~30nm、1nm~20nm、1nm~10nm、1nm~5nm的范围含,但不限于此。上述的第一、第二及第三酶组可使用市售的,也可使用从微生物等提取、根据需要纯化的。来自微生物的酶的提取及纯化可使用本领域技术人员能利用的方法而适宜实施。当作为上述第一、第二及第三酶组,使用以上所示的大肠杆菌来源的酶以外时,可以相对于对于上述大肠杆菌来源的酶确定的浓度范围而作为酶活性单位相当的浓度范围使用。也可使含上述酶的无细胞蛋白质表达系统的反应液直接与成为模板的环状dna混合而形成用于环状dna的扩增的反应混合液。无细胞蛋白质表达系统可为以含由与编码上述酶的基因的碱基序列互补的序列构成的rna的总rna(totalrna)、mrna、或者invitro转录产物等作为模板rna的无细胞翻译系统,也可为以编码各酶的基因或含编码各酶的基因的表达载体等作为模板dna的无细胞转录翻译系统。<环状dna的扩增方法>本发明涉及环状dna的扩增方法,其包括以下的工序:(1)形成成为模板的环状dna和含以下物质的反应液的反应混合物的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源,其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric))。在别的实施方式中,本发明的方法也可在上述工序(1)之前,还含对反应液进行预温育的工序。即,本发明的方法可为环状dna的扩增方法,其含以下的工序:(1-1)对含以下物质的反应液进行预温育的工序:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源;及(1-2)形成所述反应液和成为模板的环状dna的反应混合物的工序,其中所述环状dna含能与有dnaa活性的酶结合的复制起始序列(originofchromosome(oric))。预温育也可通过在例如0~40℃、10~40℃、15~37℃、或者16~30℃的范围保温5~60分钟、5~45分钟、5~30分钟、15~60分钟、15~45分钟、15~30分钟之间来进行。预温育只要是反应液的温度保持在上述的温度范围内,就也可在预温育中若干变动。不受理论限制,如本发明重复图1所示复制循环,指数扩增环状dna。在本发明中,以上述的环状dna作为模板使用而可将其扩增至少10倍、50倍、100倍、200倍、500倍、1000倍、2000倍、3000倍、4000倍、5000倍、或者10000倍。对于与反应液混合的环状dna,如在上述<环状dna>的项目中记载。在每1反应中使用的模板dna的量无特别限制,也可例如在反应开始时以10ng/μl以下,5ng/μl以下,1ng/μl以下,0.8ng/μl以下,0.5ng/μl以下,0.3ng/μl以下的浓度存在于反应液中。再者,在反应开始时,也可以每1反应1分子的环状dna作为模板存在而在扩增中使用。反应液中所含的缓冲液只要是对于在ph7~9、优选为ph8使用而言适宜的缓冲液,就无特别限制。例如,可举出tris-hcl、tris-oac、hepes-koh、磷酸缓冲液、mops-naoh、tricine-hcl等。优选的缓冲液是tris-hcl或tris-oac。缓冲液的浓度可由本领域技术人员适宜选择,不特别限定,在tris-hcl或tris-oac的情况中,可选择例如10mm~100mm、10mm~50mm、20mm的浓度。atp是指腺苷三磷酸。在反应开始时反应液中所含的atp的浓度可为例如0.1mm~3mm的范围,也可优选为0.1mm~2mm、0.1mm~1.5mm、0.5mm~1.5mm的范围。gtp、ctp及utp各自是指鸟苷三磷酸、胞苷三磷酸、及尿苷三磷酸。在反应开始时反应液中所含的gtp、ctp及utp的浓度可各自独立地为例如0.1mm~3.0mm的范围,也可优选为0.5mm~3.0mm、0.5mm~2.0mm的范围。dntp是脱氧腺苷三磷酸(datp)、脱氧鸟苷三磷酸(dgtp)、脱氧胞苷三磷酸(dctp)、及脱氧胸腺嘧啶三磷酸(dttp)的总称。在反应开始时反应液中所含的dntp的浓度可为例如0.01~1mm的范围,也可优选为0.05mm~1mm、0.1mm~1mm的范围。镁离子源是向反应液中给镁离子(mg2+)的物质。例如,可举出mg(oac)2、mgcl2、及mgso4、等。优选的镁离子源是mg(oac)2。在反应开始时反应液中所含的镁离子源的浓度可为例如在反应液中以5~50mm的范围给镁离子的浓度。碱金属离子源是向反应液中给碱金属离子的物质。作为碱金属离子,例如可举出钠离子(na+)、钾离子(k+)。作为碱金属离子源的例,可举出谷氨酸钾、天冬氨酸钾、氯化钾、醋酸钾、谷氨酸钠、天冬氨酸钠、氯化钠、及醋酸钠。优选的碱金属离子源是谷氨酸钾或醋酸钾。在反应开始时反应液中所含的碱金属离子源的浓度可在反应液中以100mm以上、优选为100mm~300mm的范围给碱金属离子的浓度,但不限于此。在与先前的申请的调和中,也可从上述的碱金属离子源的浓度除150mm。在本发明的方法中使用的反应液也可还含蛋白质的非特异吸附抑制剂或核酸的非特异吸附抑制剂。优选为,反应液也可还含蛋白质的非特异吸附抑制剂及核酸的非特异吸附抑制剂。通过蛋白质的非特异吸附抑制剂及/或核酸的非特异吸附抑制剂存在于反应液中,反应效率提升。蛋白质的非特异吸附抑制剂及/或核酸的非特异吸附抑制剂被认为通过抑制蛋白质彼此及/或蛋白质和环状dna的非特异吸附,或蛋白质及环状dna向容器表面的附着而反应效率提升。蛋白质的非特异吸附抑制剂是指与本发明的方法中的扩增反应无关系的蛋白质。作为这样的蛋白质,例如,可举出牛血清白蛋白(bsa)、溶菌酶、明胶、肝素、及酪蛋白等。蛋白质的非特异吸附抑制剂可在反应液中以0.02~2.0mg/ml的范围、优选为0.1~2.0mg/ml、0.2~2.0mg/ml、0.5~2.0mg/ml的范围含,但不限于此。核酸的非特异吸附抑制剂是指与本发明的方法中的扩增反应无关系的核酸分子或核酸类似因子。作为这样的核酸分子或核酸类似因子,例如,可举出trna(转移rna)、rrna(核糖体rna)、mrna(信使rna)、糖原、肝素、寡dna、多聚(i-c)(聚肌苷-聚胞苷)、多聚(di-dc)(聚脱氧肌苷-聚脱氧胞苷)、多聚(a)(聚腺嘌呤)、及多聚(da)(聚脱氧腺嘌呤)等。核酸的非特异吸附抑制剂可在反应液中以1~500ng/μl的范围、优选为10~500ng/μl、10~200ng/μl、10~100ng/μl的范围含,但不限于此。在与先前的申请的调和中,作为核酸的非特异吸附抑制剂选择trna时,也可从trna的浓度除50ng/μl。在本发明的方法中使用的反应液也可还含dna的稳定化因子。认为通过dna的稳定化因子存在于反应液中,dna的切断被抑制而可保护模板dna及扩增产物。由dna的稳定化因子的添加与目的产物的收率提升相关联。特别是,当模板dna是长链环状dna时,由于模板dna及扩增产物易被分解,dna的稳定化因子的添加是有益的。dna的稳定化因子不特别限定,也可为选自例如,葡萄糖、蔗糖、二甲基亚砜(dmso)、牛血清白蛋白(bsa)、二醇醚二胺四醋酸(egta)、浴铜灵二磺酸二钠(bda)、青霉胺、钛铁试剂(tiron、1,2-二羟基苯-3,5-磺酸酯)、二亚乙基三胺五醋酸(dtpa)、乙二胺四醋酸(edta)、及dps蛋白质(大肠杆菌来源)、金属硫蛋白质(人来源)。其中,dtpa、tiron、bda、dps蛋白质及bsa由于还有使环状dna扩增反应效率化的作用而特别优选的。dtpa或tiron可在反应液中以0.01mm~0.3mm、优选为0.05~0.15mm的范围含,但不限于此。bda可在反应液中以0.01~0.5mm、优选为0.05~0.3mm的范围含,但不限于此。dps蛋白质可在反应液中以0.3~3.0μm、优选为0.3~1.5μm的范围含,但不限于此。bsa可在反应液中以0.02~2.0mg/ml的范围、优选为0.1~2.0mg/ml、0.2~2.0mg/ml、0.5~2.0mg/ml的范围含,但不限于此。在本发明的方法中使用的反应液也可还含直链状dna特异性外切核酸酶或recg型解螺旋酶。优选为,反应液也可还含直链状dna特异性外切核酸酶及recg型解螺旋酶。通过直链状dna特异性外切核酸酶及/或recg型解螺旋酶存在于反应液中,有降低在扩增反应中由双链切断等发生的直链状dna的量,提升目的超螺旋产物的收率的效果。在本发明的方法中使用的反应液也可还含recg型解螺旋酶或单链dna特异性外切核酸酶。优选为,反应液也可还含recg型解螺旋酶及单链dna特异性外切核酸酶。通过recg型解螺旋酶及/或单链dna特异性外切核酸酶存在于反应液中,有降低在扩增反应中发生的低分子的副扩增产物的量,提升目的超螺旋产物的收率的效果。在本发明的方法中使用的反应液也可还含直链状dna特异性外切核酸酶或单链dna特异性外切核酸酶。优选为,反应液也可还含直链状dna特异性外切核酸酶及单链dna特异性外切核酸酶。通过直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶存在于反应液中,有降低在扩增反应中由双链切断等发生的直链状dna的量,提升目的超螺旋产物的收率的效果。直链状dna特异性外切核酸酶是从直链状dna的5’末端或3’末端依次水解的酶。直链状dna特异性外切核酸酶只要是有从直链状dna的5’末端或3’末端依次水解的活性,其种类或生物学来源就无特别限制。例如,可使用recbcd、λ外切核酸酶、外切核酸酶iii、外切核酸酶viii、t5外切核酸酶、t7外切核酸酶、及plasmid-safetmatp-dependentdna酶(epicentre)等。优选的直链状dna特异性外切核酸酶是recbcd。直链状dna外切核酸酶可在反应液中以0.001~1.0u/μl、优选为0.005u~1.0u/μl、0.01~1.0u/μl、0.05~1.0u/μl、或者0.1~1.0u/μl的范围含,但不限于此。对于直链状dna外切核酸酶的酶活性单位(u)是以在37℃、30分钟的反应中对于使直链状dna的1nmol的脱氧核糖核苷酸成为酸可溶性而言必要的酶量作为1u的单位。recg型解螺旋酶被认为是延伸反应的终结时解除复制叉彼此可冲突的副的dna结构的解螺旋酶的酶。recg型解螺旋酶只要是有与大肠杆菌来源的recg同样的活性,其生物学来源就无特别限制,可适宜地使用例如大肠杆菌来源的recg。大肠杆菌来源的recg可作为单体而在反应液中以100nm~800nm的范围、优选为100nm~500nm、100nm~400nm、100nm~300nm的范围含,但不限于此。recg型解螺旋酶可以对于上述大肠杆菌来源的recg确定的浓度范围内作为酶活性单位相当的浓度范围使用。单链dna特异性外切核酸酶是依次水解单链dna的5’末端或3’末端的核苷酸的酶。单链dna特异性外切核酸酶只要是有依次水解单链dna的5’末端或3’末端的核苷酸的活性,其种类或生物学来源就无特别限制。例如,可使用外切核酸酶i(exoi)、recj、外切核酸酶t等。优选的单链dna特异性外切核酸酶是exoi。单链dna特异性外切核酸酶可在反应液中以0.1~1.0u/μl的范围、优选为0.15~1.0u/μl、0.2~1.0u/μl、或者0.2~0.5u/μl的范围含,但不限于此。对于exoi的酶活性单位(u)是以在37℃、30分钟的反应中使单链dna的10nmol的脱氧核糖核苷酸成为酸可溶性而言必要的酶量作为1u的单位。对于recj的酶活性单位(u)是以37℃、30分钟的反应中使单链dna的0.05nmol的脱氧核糖核苷酸成为酸可溶性而言必要的酶量作为1u的单位。在本发明的方法中使用的反应液也可还含铵盐。作为铵盐的例,可举出硫酸铵、氯化铵、及醋酸铵。特别是优选的铵盐是硫酸铵或醋酸铵。铵盐可在反应液中以0.1mm~100mm的范围、优选为0.1mm~50mm、1mm~50mm、1mm~20mm的范围含,但不限于此。作为第二酶组之一,作为有dna连接酶活性的酶,使用大肠杆菌来源的dna连接酶时,作为其辅因子的nad(烟酰胺腺嘌呤二核苷酸)含在反应液中。nad可在反应液中以0.01mm~1.0mm的范围、优选为0.1mm~1.0mm、0.1mm~0.5mm的范围含,但不限于此。在本发明的方法中使用的反应液也可还含还原剂。作为优选的还原剂的例,可举出dtt、β-巯基乙醇(2-巯基乙醇)、三(2-羧基乙基)膦(tcep)及谷胱甘肽。优选的还原剂是dtt。还原剂也可以1.0mm~15.0mm的浓度,优选为以2.0mm~10.0mm、4.0mm~8.0mm的浓度含在反应液中。在本发明的方法中使用的反应液也可还含用于再生atp的酶及底物。作为atp再生系统的酶和底物的组合,可举出肌酸激酶和肌酸磷酸、及丙酮酸激酶和磷酸烯醇丙酮酸。作为atp再生系统的酶,可举出肌激酶。优选的atp再生系统的酶和底物的组合是肌酸激酶及肌酸磷酸。对于反应液中所含的第一、第二、及第三酶组,如在上述<第一、第二及第三酶组>的项目中记载。在某实施方式中,在本发明的方法中使用的第一酶组可含具有dnaa活性的酶、1种以上的核样体蛋白质、有dna促旋酶活性的酶或酶组、单链dna结合蛋白质(single-strandbindingprotein(ssb))、有dnab型解螺旋酶活性的酶、有dna解螺旋酶装载体活性的酶、有dna引发酶活性的酶、有dna夹活性的酶、及有dna聚合酶iii*活性的酶或酶组的组合。其中,1种以上的核样体蛋白质可为ihf或hu,有dna促旋酶活性的酶或酶组可为由gyra及gyrb构成的复合物,有dnab型解螺旋酶活性的酶可为dnab解螺旋酶,有dna解螺旋酶装载体活性的酶可为dnac解螺旋酶装载体,有dna引发酶活性的酶可为dnag引发酶,有dna夹活性的酶可为dnan夹,进而,有dna聚合酶iii*活性的酶或酶组可为含dnax、hola、holb、holc、hold、dnae、dnaq、及hole之任一者的酶或酶组。在别的实施方式中,在本发明的方法中使用的第二酶组可含具有dna聚合酶i活性的酶及有dna连接酶活性的酶的组合。或者,第二酶组可含具有dna聚合酶i活性的酶、有dna连接酶活性的酶、及有rna酶h活性的酶的组合的。在另外别的实施方式中,在本发明的方法中使用的第三酶组可含具有拓扑异构酶iii活性的酶及/或有拓扑异构酶iv活性的酶。或者,第三酶组可含具有拓扑异构酶iii活性的酶及有recq型解螺旋酶活性的酶的组合。或者另外,第三酶组也可为有拓扑异构酶iii活性的酶、有recq型解螺旋酶活性的酶、及有拓扑异构酶iv活性的酶的组合。本发明的方法也可作为工序(2),还含将上述反应混合物在指定的温度范围保温的工序。指定的温度范围只要是可使dna复制反应进行,就无特别限制,可为例如作为dna聚合酶的最适温度的20℃~80℃、25℃~50℃、或者25℃~40℃的范围。在指定的温度范围内的保温在反应中容许该指定的温度范围内的温度变化或温度变动。在优选的实施方式中,上述工序(2)也可为将上述反应混合物在等温条件下保温的工序。作为等温条件,只要是可使dna复制反应进行,就无特别限制,可设为例如作为dna聚合酶的最适温度的20℃~80℃的范围中所含的一定的温度,可设为25℃~50℃的范围中所含的一定的温度,可设为25℃~40℃的范围中所含的一定的温度,可设为30℃左右。在本说明书中在“等温条件下保温”、“使在等温反应”的用语是指保持在相对于反应中设定的温度而±7℃、±5℃、±3℃、或者±1℃的温度范围内。保温时间可对应于作为目的的环状dna的扩增产物的量而适宜设定,可设为例如1~24小时。或者,本发明的方法也可作为工序(2),还含将上述反应混合物在重复30℃以上的温育及27℃以下的温育的温度循环下温育的工序。在30℃以上的温育只要是含oric的环状dna的复制起始可能的温度范围,就无特别限定,可为例如30~80℃、30~50℃、30~40℃、37℃。在30℃以上的温育不特别限定,也可为每1个循环10秒钟~10分钟。在27℃以下的温育只要是复制起始被抑制,dna的延伸反应进行的温度,就无特别限定,可为例如10~27℃、16~25℃、24℃。在27℃以下的温育不特别限定,优选对应于扩增的环状dna的长度而设定,也可为例如对于1个循环,每1000个碱基1~10秒钟。温度循环的循环数不特别限定,也可为10~50个循环、20~40个循环、25~35个循环、30个循环。在某实施方式中,工序(2)也可在油包水滴型乳液内进行。油包水滴型乳液可通过向工序(1)中形成的反应混合物添加矿物油及表面活性剂而使混合来调制。矿物油及表面活性剂的种类及量可由本领域技术人员适宜选择。本发明的方法也可在工序(2)之后,还含用不含第一至第三酶组的反应液稀释为五倍以上之后,再保温的工序。由酶组的稀释通过抑制新的复制起始的一方而进行途中的复制延伸、环连体形成、分离反应由残留酶的效果而继续进行。另外,即使是反应中切口进入等而发生的副产物,也能在此过程中由残留连接酶等的效果修复。从而,特异性地导致自扩增中间体或副产物向最终产物的迁移,可期待目的超螺旋结构的环状dna的收率提升。本发明的方法也可在工序(2)之后,还含用直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶处理的工序。通过用直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶处理,可分解作为在扩增反应中发生的副产物的直链状dna而除去。直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶的种类及使用的量也可为如上所述的。由直链状dna特异性外切核酸酶及/或单链dna特异性外切核酸酶的处理也可例如于25℃~40℃进行30分钟~3小时。本发明的方法也可在工序(2)之后,还含用缺口修复酶处理的工序。缺口修复酶是修复在双链dna中缺少1个或多个连续的核苷酸的状态的缺口、或在双链dna中邻接的核苷酸间的磷酸二酯键被切断的状态的切口,作为完全的双链超螺旋dna的酶组。通过用缺口修复酶处理,有修复在扩增反应中作为副产物发生的缺口或切口进入的dna,提升目的超螺旋产物的收率的效果。缺口修复酶只要是可修复双链dna的缺口或切口的酶组,其种类或生物学来源就无特别限制。例如,可使用外切核酸酶iii、dna聚合酶i、dna连接酶、有dna促旋酶活性的酶或酶组的组合。有外切核酸酶iii活性的酶可以5~100mu/μl的浓度使用,但不限于此。对于外切核酸酶iii的酶活性单位(u)是以37℃、30分钟的反应中使双链dna的1nmol的脱氧核糖核苷酸成为酸可溶性而言必要的酶量作为1u的单位。dna聚合酶i、dna连接酶、有dna促旋酶活性的酶或酶组可以对于各自前述的第一或第二酶组确定的浓度使用,但不限于此。由缺口修复酶的处理也可例如于25~40℃进行5~120分钟、优选为10~60分钟。本发明的方法也可在工序(2)之后,包括对应于目的而纯化环状dna的扩增产物的工序。环状dna的纯化可使用本领域技术人员能利用的方法而适宜实施。可将使用本发明的方法而扩增的环状dna直接或者适宜纯化反应后的反应混合物的在转化等的其后的目的中使用。<环状dna的扩增用组合物及试剂盒>本发明涉及环状dna的扩增用组合物,其含:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源。本发明的组合物也可还含选自蛋白质的非特异吸附抑制剂、核酸的非特异吸附抑制剂、直链状dna特异性外切核酸酶、recg型解螺旋酶、单链dna特异性外切核酸酶、铵盐、nad、还原剂、dna的稳定化因子、以及,atp再生系统的酶及底物的组合的1种以上的成分。对于本发明的组合物中所含的成分的具体性的成分及浓度而言,如在上述<环状dna>、<第一、第二、第三酶组>、<环状dna的扩增方法>的项目中记载。另外,本发明还涉及环状dna的扩增用试剂盒,其含下列物质的组合:催化环状dna的复制的第一酶组;催化冈崎片段连接反应而合成形成环连体的2个姊妹环状dna的第二酶组;催化2个姊妹环状dna的分离反应的第三酶组;缓冲液;atp;gtp、ctp及utp;dntp;镁离子源;及碱金属离子源。本发明的试剂盒可在1个试剂盒中含全部上述的构成品,另外,只要是用于在本发明的方法中利用的目的的试剂盒,就也可不含上述的构成品的一部分。在是不含上述的构成品的一部分的试剂盒时,实施者可向所述试剂盒追加扩增时必要的成分而实施本申请发明的扩增方法。本发明的试剂盒也可还含有含选自蛋白质的非特异吸附抑制剂、核酸的非特异吸附抑制剂、直链状dna特异性外切核酸酶、recg型解螺旋酶、单链dna特异性外切核酸酶、铵盐、nad、还原剂、dna的稳定化因子、以及,atp再生系统的酶及底物的组合的1种以上的成分的追加的构成品。本发明的试剂盒也可还另外为了扩增反应后的处理而含有含选自直链状dna特异性外切核酸酶、单链dna特异性外切核酸酶、及缺口修复酶的1以上的成分的追加的构成品。追加的构成品可为作为1个试剂盒含在本发明的试剂盒中,或也可作为以与本发明的试剂盒一同使用作为前提的别的试剂盒提供。对于本发明的试剂盒中所含的各构成品的具体性的成分及浓度而言,如在上述<环状dna>、<第一、第二、第三酶组>、<环状dna的扩增方法>的项目中记载。本发明的试剂盒可为含将上述构成品的混合物包装为1个的,也可为含将上述构成品个别地或者各数种综合而混合的个别地包装的。本发明的试剂盒也可还含记载了用于实施本发明的环状dna的扩增方法的指示的说明书。在所述说明书中,也可作为说明记载上述<环状dna>、<第一、第二、第三酶组>、<环状dna的扩增方法>的项目中记载的事项。【实施例】以下,基于实施例具体说明本发明。再者,本发明不限于下述实施例中记载的范围。【实施例1:环状dna的扩增】<材料和方法>向表1所示的组成的反应液添加模板dna而在冰上混合之后,在30℃的温育器中保温1小时、2小时、或者3小时。使每1个反应的总容量成为10μl。在30℃的反应后,对反应产物进行琼脂糖凝胶电泳(0.5%1×tae、150v、100分钟、14℃)之后,使用sybrgreen(宝生物株式会社)检测dna。【表1】表中,ssb表示大肠杆菌来源ssb、ihf表示大肠杆菌来源ihfa及ihfb的复合物、dnag表示大肠杆菌来源dnag、dnan表示大肠杆菌来源dnan、poliii*表示作为由大肠杆菌来源dnax、hola、holb、holc、hold、dnae、dnaq、及hole组成的复合物的dna聚合酶iii*复合物、dnab表示大肠杆菌来源dnab、dnac表示大肠杆菌来源dnac、dnaa表示大肠杆菌来源rna酶h、连接酶表示大肠杆菌来源dna连接酶、poli表示大肠杆菌来源dna聚合酶i、gyra表示大肠杆菌来源gyra、gyrb表示大肠杆菌来源gyrb、topoiv表示大肠杆菌来源parc及pare的复合物、topoiii表示大肠杆菌来源拓扑异构酶iii、recq表示大肠杆菌来源recq。ssb从ssb的大肠杆菌表达株,由含硫酸铵沉淀及离子交换柱层析的工序纯化、调制。ihf从ihfa及ihfb的大肠杆菌共表达株,由含硫酸铵沉淀及亲和性柱层析的工序纯化、调制。dnag从dnag的大肠杆菌表达株,由含硫酸铵沉淀、阴离子交换柱层析、及凝胶过滤柱层析的工序纯化、调制。dnan从dnan的大肠杆菌表达株,由含硫酸铵沉淀及阴离子交换柱层析的工序纯化、调制。poliii*从dnax、hola、holb、holc、hold、dnae、dnaq及hole的大肠杆菌共表达株,由含硫酸铵沉淀、亲和性柱层析、及凝胶过滤柱层析的工序纯化、调制。dnab、dnac从dnab及dnac的大肠杆菌共表达株,由含硫酸铵沉淀、亲和性柱层析、及凝胶过滤柱层析的工序纯化、调制。dnaa从dnaa的大肠杆菌表达株,由含硫酸铵沉淀、透析沉淀、及凝胶过滤柱层析的工序纯化、调制。gyra、gyrb从gyra的大肠杆菌表达株和gyrb的大肠杆菌表达株的混合物,由含硫酸铵沉淀、亲和性柱层析、及凝胶过滤柱层析的工序纯化、调制。topoiv从parc的大肠杆菌表达株和pare的大肠杆菌表达株的混合物,由含硫酸铵沉淀、亲和性柱层析、及凝胶过滤柱层析的工序纯化、调制。topoiii从topoiii的大肠杆菌表达株,由含硫酸铵沉淀及亲和性柱层析的工序纯化、调制。recq从recq的大肠杆菌表达株,由含硫酸铵沉淀、亲和性柱层析、及凝胶过滤柱层析的工序纯化、调制。rna酶h、连接酶、poli使用市售的大肠杆菌来源的酶(宝生物株式会社)。作为模板dna,使用9.6kb的环状dna(具有复制起始序列oric的环状dna、卡那霉素抗性(km))、80kb的长链环状dna(具有复制起始序列oric的环状dna、卡那霉素抗性(km))、200kb的长链环状dna(具有复制起始序列oric的环状dna、卡那霉素抗性(km))、或者由试管内连接环化的dna。9.6kb的环状dna及80kb、200kb的长链环状dna由大肠杆菌细胞内重组反应调制。具体而言,使用表达λ噬菌体的重组蛋白质组的大肠杆菌,由细胞内重组反应调制含卡那霉素抗性盒和含大肠杆菌染色体之中oric的区域的期望的长度的环状dna。由试管内连接的环化dna的调制方法示于图2。具体而言,尽管具有复制起始序列oric和卡那霉素抗性(km),通过使pcr片段(2.3kb)与有dnaa基因及dnan基因序列的pcr片段(以下记载为dnaa-dnan片段)(2.6kb)由gibson组装法反应而连接。2个pcr片段经附加在两端的20碱基的相同序列(记载为h1、h2)而连接,变得环状结构。反应通过向gibson组装mastermix(neb公司)混合上述2种pcr片段,于50℃反应15分钟而进行。反应后,以反应溶液0.1μl分(反应溶液的稀释10倍溶液1μl分)作为模板dna,直接添加到10μl的扩增反应系统(表1的组成)中,于30℃反应1小时。<结果1>以9.6kb的环状dna(0.08ng、作为环状分子数而约107个)作为模板使用时由sybrgreen的扩增产物的检测结果示于图3。尽管也见到副产物(反应中间产物),但可确认超螺旋结构的环状dna扩增产物(以黑框表示)。通过直接使用反应后的溶液而转化大肠杆菌dh5α株,用含有卡那霉素的琼脂培养基培养,测量集落数而求出环状dna的扩增量的结果示于表2。作为对照,使用保温时间0小时的反应溶液。【表2】保温时间0小时3小时转化体(集落数)95.4×104可由本发明的方法,以9.6kb的环状dna作为环状dna而扩增大概6,000倍。<结果2>以80kb及200kb的长链环状dna作为模板使用时由sybrgreen的扩增产物的检测结果示于图4。可确认超螺旋结构的环状dna扩增产物(以黑框或箭头表示)。得知由本发明的方法,即使在以80kb或200kb的长而大的环状dna作为模板使用时,也良好地得到了扩增产物。<结果3>以由试管内连接环化的dna作为模板使用时由sybrgreen的扩增产物的检测结果示于图5。得知由本发明的方法,即使以由试管内连接环化的dna作为模板使用时,也良好地得到了扩增产物。通过直接使用反应后的溶液而转化大肠杆菌dh5α株,用含有卡那霉素的琼脂培养基培养,测量集落数而求出环状dna的扩增量的结果示于表3。作为对照,使用不进行扩增反应的样品。【表3】扩增反应-+转化体(集落数)3.2×1026.6×105得知使用gibson组装法环化的dna由本发明的方法,作为环状分子扩增为大概2,000倍。【实施例2:来自少数的模板分子的环状dna的扩增】使用实施例1中记载的9.6kb的环状dna,与实施例1同样地进行扩增反应。(2-1)扩增效率的探讨通过向扩增反应液(实施例1、表1)10μl作为环状分子加9.6kb的环状dna至含1~1000个分子,于30℃保温3小时来进行扩增反应。对于反应物而进行0.5%琼脂糖凝胶电泳,用sybrgreen(宝生物株式会社)染色,检测扩增dna(图6a)。另外,将扩增产物的总dna量由picogreen检测试剂盒(thermofisher公司)定量(图6b:picogreen法)。通过将扩增产物直接转化大肠杆菌,求出卡那霉素抗性集落数来定量作为环状dna分子的扩增量(图6b:转化法)。从定量结果求出相对于初期dna量的扩增程度(扩增)、示于坐标图。从上述的结果得知,以区区3小时的等温反应能将作为模板dna1分子的环状dna扩增至约1011分子(约1,000亿倍的扩增)。(2-2)倍增时间的探讨通过向扩增反应液(实施例1、表1)80μl加上述的环状dna,于30℃保温来进行扩增反应。环状dna以每1μl反应液成为105分子的方式加。通过经时采样,将样品直接转化大肠杆菌,求出卡那霉素抗性集落数来对扩增的环状dna分子数进行定量(图7)。由上述的结果确认到9.6kb的环状dna分子的倍增时间是约5分钟。【实施例3:从混合物扩增单一的环状dna克隆】从实施例1中记载的9.6kb的环状dna及12.0kb的环状dna(具有复制起始序列oric的环状dna、卡那霉素抗性(km))的混合物进行单一的环状dna克隆的扩增。12.0kb的环状dna由大肠杆菌细胞内重组反应调制。具体而言,使用表达λ噬菌体的重组蛋白质组的大肠杆菌,由细胞内重组反应调制含由oric和卡那霉素抗性基因构成的盒和大肠杆菌染色体的一部分的区域的期望的长度的环状dna。通过将上述2种环状dna的混合物在反应液中稀释为成为各15分子或各1.5分子而加到扩增反应液(实施例1、表1)10μl中,于30℃保温6小时来进行扩增反应。对于反应物而进行0.5%琼脂糖凝胶电泳,用sybrgreen(宝生物株式会社)染色,检测扩增dna(图8)。结果,在环状dna被稀释至1.5分子的反应液中,在各反应样品全部中,仅任一方的克隆被扩增。这表明,即使模板dna是混合物,通过将其在反应液中稀释至1分子水平,单一的环状dna克隆的扩增也是可能的。【实施例4:传代扩增】使用lacz环状dna而对于环状dna的传代扩增而进行试验。lacz环状dna(9.0kb)通过使含oric的双链dna片段(1.0kb)、含卡那霉素抗性基因(km)的双链dna片段(4.6kb)及含lacz(β-半乳糖苷酶)基因的双链dna片段(3.4kb)连接而调制。通过向扩增反应液(实施例1、表1)10μl加lacz环状dna至含1,000个分子,于30℃保温3小时来进行扩增反应,以其作为1传代。通过将在之前的传代数的扩增反应物稀释105倍,将1μl其添加到新的扩增反应液中而同样地反应而作为此后的传代扩增。重复此传代扩增至10次。对于每传代的扩增产物而进行0.5%琼脂糖凝胶电泳,用sybrgreen(宝生物株式会社)染色,检测(图9)。另外,使其扩增产物的一部分大肠杆菌转化,由卡那霉素抗性集落数对dna扩增程度进行定量,由此值算出重复指数扩增几个世代而显示为总世代数。结果,得知即使在10次的传代后环状dna的扩仍增有效进行。如实施例2(图7)的结果所示,环状dna某程度扩增,则由底物或酶的枯渴,扩增速度达到顶点。一方面,本实施例的结果表明,通过将扩增反应物的一部分用新的反应液传代,能半永久地重复环状dna的指数扩增。即,本发明的方法是能如细胞的传代增殖一样进行环状dna的扩增的方法。【实施例5:扩增反应中的复制错误发生率】由于在实施例4中的模板的环状dna中含lacz基因,将用此环状基因转化的大肠杆菌在x-gal板上培养,则由于lacz基因正常表达的集落(lacz+)可分解x-gal而呈蓝色,由于因复制错误导致的变异导入而lacz基因变得无法正常发挥功能的集落(lacz-)无法分解x-gal而呈白色。即,可由用此环状基因转化的大肠杆菌在x-gal板上所呈的色彩判断扩增的环状dna中的复制错误。将各传代样品的扩增反应物直接转化大肠杆菌,在x-gal板上培养而求出lacz-出现率。从此lacz-出现率和实施例4中求出的总世代数,根据barnes的方法(barneswmgene.1992、112、29-35)算出复制循环每1世代的错误发生率。结果示于以下的表4。【表4】传代数总世代数lacz-集落出现率(%)错误发生率(每碱基)125<0.045<3.7×10-8244<0.074<3.4×10-851060.11.9×10-8102010.0930.93×10-8上述的结果表明,复制错误是对于1亿碱基而1个位置左右(每碱基平均1.4×10-8错误)。这是与细胞内(不具有错配修复系统的株)的变异率同程度,是taq聚合酶的约1万倍的正确性。【实施例6:外切核酸酶及recg的追加】使用实施例1中记载的80kb的环状dna,探讨向扩增反应液还添加recg型解螺旋酶及直链状dna特异性外切核酸酶而进行扩增反应时的效果。80kb的环状dna如实施例1所示调制。作为recg型解螺旋酶,使用recg。recg从recg的表达大肠杆菌发的株,由含硫酸铵沉淀、亲和性柱层析的工序纯化、调制。作为直链状dna特异性外切核酸酶,使用作为市售的外切核酸酶的plasmid-safetmatp-dependentdna酶(epicentre)。单位数根据制造方的记载。通过将向实施例1的表1中所示的反应组成加80kb的环状dna至成为0.8pg/μl或8pg/μl、加recg至成为0nm、100nm、300nm、或者1000nm、及加直链状dna特异性外切核酸酶至成为0u/μl或0.2u/μl的扩增反应液(10μl)于30℃保温24小时来进行扩增反应。对于反应物而进行0.5%琼脂糖凝胶电泳(1×tae缓冲液、150v、100分钟),用sybrgreen(宝生物株式会社)染色,检测扩增dna。结果,观察到由recg及直链状dna特异性外切核酸酶的添加降低作为dna的切断等导致的副产物的直链状dna的生成,目的超螺旋结构的环状dna扩增产物的生成量的提升(图10)。【实施例7:各种条件探讨】表示对于反应液的各成分进行条件探讨的结果。1.方法作为成为模板的环状dna,使用8.0kb的环状dna。8.0kb的环状dna向m13mp18质粒载体插入oric片段而制成。对于条件a~r,向表5中所示的扩增反应液加8.0kb的环状dna至成为终浓度8.0ng/μl或0.8ng/μl,于30℃反应1小时。向反应液添加[α-32p]datp,在dna复制反应后,用液体闪烁计数器测量摄入到模板dna的dntp量。dntp摄入量算出每25μl反应液的值。以终浓度8.0ng/μl的环状dna作为模板,100%复制反应进行时(1轮的复制),摄入600pmol的dntp。反应液的一部分在琼脂糖凝胶电泳后,用bas成像板检测32p摄入产物,确认目的超螺旋结构产生。对于条件s,向表5中所示的扩增反应液加8.0kb的环状dna至成为终浓度8.0ng/μl或0.8ng/μl,于30℃反应2小时。对于反应物而进行0.5%琼脂糖凝胶电泳(1×tae缓冲液、150v、100分钟),用sybrgreen(宝生物株式会社)染色,检测扩增dna。【表5-1】【表5-2】【表5-3】2.结果(1)条件a通过加第一、第二及第三酶组而使反应,发现导致数轮的复制循环的重复。但是确证,由于每经一次复制循环就反应的底物蛋白质越发变得不足,复制循环在至4轮停滞(图11)。(2)条件b探讨通过减少反应开始的模板dna量而是否可使复制循环数提升。对于反应开始时的模板dna量,以8ng/μl及0.8ng/μl探讨。结果,尽管在反应开始时的模板dna量是8ng/μl时见到复制反应,将模板dna量减少到0.8ng/μl,则复制效率显著地被抑制,观察不到dna的扩增(图12)。这表明,为了使复制循环数提升,不是仅使模板dna量减少即可的,含反应组成的各种成分的量的条件探讨是必要的。(3)条件c探讨在反应组成中的gtp、ctp及utp的量。对于反应开始时的gtp、ctp及utp的浓度而探讨0.2mm、0.5mm、1.0mm及2.0mm。结果示于图13。(4)条件d探讨在反应组成中的ihf的量。对于反应开始时的ihf的浓度而探讨0nm、10nm、20nm、40nm、100nm、及200nm。结果示于图14。(5)条件e探讨在反应组成中的topoiv的量。对于反应开始时的topoiv的浓度而探讨0nm、1nm、2nm、5nm、10nm、及20nm。结果示于图15。(6)条件f探讨在反应组成中的dna促旋酶的量。对于反应开始时的gyra-gyrb复合物的浓度而探讨0nm、10nm、25nm、50nm、100nm、及200nm。结果示于图16。(7)条件g探讨在反应组成中的dna聚合酶iii*的量。对于反应开始时的poliii*的浓度而探讨0nm、1nm、2nm、5nm、及10nm。结果示于图17。(8)条件h探讨在反应组成中的碱金属离子源的量。对于反应开始时的谷氨酸钾的浓度而探讨50mm及150mm。结果示于图18。(9)条件i探讨在反应组成中的蛋白质的非特异吸附抑制剂及/或核酸的非特异吸附抑制剂的量。探讨反应组成中不含trna而含0.1mg/mlbsa的条件、含20ng/μltrna及0.1mg/mlbsa的条件、不含trna而含bsa0.5mg/ml的条件。结果示于图19。(10)条件j探讨在反应组成中的有dnaa活性的酶的量。对于反应开始时的dnaa的浓度而探讨0nm、5nm、10nm、20nm、40nm、100nm、及200nm。结果示于图20。(11)条件k探讨在反应组成中的有dna连接酶活性的酶的量。对于反应开始时的连接酶的浓度而探讨0nm、2nm、5nm、10nm、20nm、及50nm。结果示于图21。(12)条件l探讨在反应组成中的单链dna结合蛋白质(ssb)的量。对于反应开始时的ssb的浓度而探讨0nm、10nm、20nm、50nm、100nm、200nm、及500nm。结果示于图22。(13)条件m探讨在反应组成中的有dna聚合酶i活性的酶的量。对于反应开始时的poli的浓度而探讨0nm、2nm、5nm、10nm、20nm、及50nm。结果示于图23。(14)条件n探讨在反应组成中的有dnab型解螺旋酶活性的酶及有dna解螺旋酶装载体活性的酶的量。对于反应开始时的dnab-dnac复合物的浓度而探讨0nm、5nm、10nm、20nm、及40nm。结果示于图24。(15)条件o探讨在反应组成中的有rna酶h活性的酶的量。对于反应开始时的rna酶h的浓度而探讨1nm、3nm、及10nm。结果示于图25。(16)条件p对于条件p而探讨将反应开始时的模板dna量设为8ng/μl、0.8ng/μl、及0.27ng/μl而进行扩增反应。结果示于图26。在条件p中,在模板dna的量是0.8ng/μl时也可有效扩增。再者,将模板dna的量减少至0.27ng/μl时也可有效扩增。作为dna合成量,可确认到超100倍而扩增。(17)条件q对于反应组成中的第三酶组的酶的组成及量而进行探讨。作为第三酶组,使用topoiv、topoiii、及recq。探讨各酶的浓度如图27所示。结果示于图27。(18)条件r对于条件r而探讨dna促旋酶的量。对于反应开始时的gyra-gyrb复合物的浓度而探讨0nm、10nm、25nm、50nm、及150nm。结果示于图28。(19)条件s作为条件s,变更trna、nad、硫酸铵(as)、ihf、ssb、topoiv的浓度而探讨环状dna的扩增。探讨各成分的浓度如图29所示。结果示于图29。【实施例8:缓冲液组成的改良】对于表1中所示的反应缓冲液的组成而进一步进行条件探讨。具体而言,使用0.5pm实施例1中记载的200kb环状dna,向反应缓冲液的组成加变更之外与实施例1同样地进行扩增反应。(1)二硫苏糖醇(dtt)的量的改良对于dtt,在实施例1中设为8mm的浓度时,变更为4mm而进行扩增反应。结果,确认即便使dtt的量减半,环状dna的扩增反应仍进行。(2)碱金属离子源的探讨对于表1的反应缓冲液的组成而使dtt变更为4mm的同时,不含碱金属离子源的反应缓冲液、及,作为碱金属离子源代替谷氨酸钾而使用含150mm醋酸钾的反应缓冲液,进行环状dna的扩增反应。结果示于图30。在从0.5pm以下的低浓度扩增长链环状dna时,发生低分子的副产物被扩增,变得无法确认到作为目的扩增产物的超螺旋的产生的问题。但是,通过使谷氨酸钾或醋酸钾等的碱金属离子源含在反应缓冲液,即使是在从0.5pm的低浓度扩增长链环状dna时,也可良好地确认作为目的产物的超螺旋的扩增。在往后的实验中,使用以下所示的组成的反应缓冲液。【表6】反应缓冲液tris-hcl(ph8.0)20mm二硫苏糖醇4mm醋酸钾150mmmg(oac)210mm肌酸磷酸4mmrntps各自1mmnad0.25mm硫酸铵10mmtrna50ng/μldntps各自0.1mm牛血清白蛋白(bsa)0.5mg/ml肌酸激酶20ng/μl(3)缓冲剂的探讨对于表6的缓冲液的组成,将20mmtris-hcl(ph8.0)变更为20mmtris-oac(ph8.0)而进行环状dna的扩增反应。结果,在使用20mmtris-oac(ph8.0)时,也观察到与使用20mmtris-hcl(ph8.0)时同样的扩增产物。(4)二硫苏糖醇(dtt)的替代物的探讨对于表6的缓冲液组成,将4mmdtt变更为4mm2-巯基乙醇(2-me)或4mm三(2-羧基乙基)膦(tcep)而进行环状dna的扩增反应。结果,在使用2-me及tcep之任一者时,也观察到与使用dtt时同样的扩增产物。(5)硫酸铵的替代物的探讨对于表6的缓冲液组成,将10mm硫酸铵变更为10mm醋酸铵而进行环状dna的扩增反应。结果,在使用醋酸铵时,也观察到与使用硫酸铵时同样的扩增产物。【实施例9:由反应液的预温育的扩增效率化】探讨在扩增反应前进行预温育时的效果。作为模板dna,使用实施例1中记载的200kb环状dna。在冰上调制含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液。于0℃、16℃、或者30℃,各自进行0、5、15或30分钟预温育。其后,向反应液添加模板dna至终浓度成为0.05pm,在30℃的温育器中保温3小时。反应后,将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图31。于16℃或30℃进行预温育时,确认到作为目的产物的超螺旋的产生增加。扩增反应前的预温育即使在长链环状dna的自低浓度的扩增时,也在作为目的扩增产物的超螺旋的产生变多的点有效。【实施例10:recg及recj的追加】探讨向扩增反应液还添加recg型解螺旋酶及单链dna特异性外切核酸酶而进行扩增反应时的效果。作为模板dna,使用实施例1中记载的200kb环状dna。作为recg型解螺旋酶,使用recg。recg使用与实施例6同样地调整的。作为单链dna特异性外切核酸酶,使用recj。recj获自neb公司。通过将向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加200kb环状dna至成为0.5pm(67pg/μl)、加recg至成为0nm或100nm、加recj至成为0u/μl或0.5u/μl的扩增反应液(10μl)于30℃保温3小时或25小时来进行扩增反应。反应后,将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图32。由recg及recj的添加降低低分子的副扩增产物的纯化,观察到目的超螺旋结构的环状dna扩增产物的生成量的提升。在扩增反应中的recg型解螺旋酶及单链dna特异性外切核酸酶的添加特别是作为解除从低浓度扩增长链环状dna时低分子的副产物被扩增的问题的手段有效。【实施例11:recbcd及exoi的追加】探讨向扩增反应液还添加直链状dna特异性外切核酸酶及单链dna特异性外切核酸酶而进行扩增反应时的效果。作为模板dna,使用实施例1中记载的200kb环状dna。作为直链状dna特异性外切核酸酶,使用recbcd。recbcd获自neb公司。作为单链dna特异性外切核酸酶,使用exoi。exoi获自neb公司。通过将向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加200kb环状dna至成为0.5pm(67pg/μl)、加recbcd至成为0、1.5、5.0、15.0、或者50.0mu/μl、加exoi至成为200mu/μl的扩增反应液(10μl)于30℃保温20小时来进行扩增反应。反应后,将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图33。观察到由recbcd及exoi的添加降低作为dna的切断等导致的副产物的直链状dna的生成,目的超螺旋结构的环状dna扩增产物的生成量的提升。在扩增反应中的直链状dna特异性外切核酸酶及单链特异性外切核酸酶的添加特别是作为解除从低浓度扩增长链环状dna时直链状dna的副产物被扩增的问题的手段有效。【实施例12:由反应后处理-稀释再保温的最终产物增加和再保温时的由recbcd及exoi的直链状dna的除去】探讨通过在扩增反应后进行稀释再保温处理而能否除去副产物。再者,探讨通过在扩增反应后用直链状dna特异性外切核酸酶及/或单链特异性外切核酸酶处理而能否除去副产物。作为模板dna,使用实施例1中记载的200kb环状dna。作为直链状dna特异性外切核酸酶,使用recbcd,作为单链dna特异性外切核酸酶,使用exoi。recbcd及exoi与实施例10同样地得到。通过将向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加200kb环状dna至成为0.5pm(67pg/μl)的扩增反应液(10μl)于30℃保温23小时来进行扩增反应。将扩增反应后的反应液用从表6除肌酸激酶和牛血清白蛋白的组成的反应缓冲液稀释为1/5,(i)直接于30℃再保温1小时、(ii)添加200mu/μl的recbcd而于30℃1小时再保温、或者(iii)添加200mu/μl的recbcd及添加200mu/μl的exoi而于30℃1小时再保温。将反应产物与稀释再保温前的产物一同,与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图34。仅通过将扩增反应后的反应液稀释再保温,也可检测到目的超螺旋结构的环状dna。再者,可在直链状dna特异性外切核酸酶及/或单链特异性外切核酸酶存在下,除去作为副产物的直链状dna。稀释再保温处理作为促进产物中的扩增中间体的复制延伸或分离反应,提高作为最终产物的超螺旋dna的产生量的手段有效。再者,再保温时由直链状dna特异性外切核酸酶及单链特异性外切核酸酶附处理特别是作为可除去从低浓度扩增长链环状dna时作为副产物发生的直链状dna的手段有效。【实施例13:由反应后处理-缺口修复(gr)酶的单链缺口的修复】扩增反应后,通过用缺口修复(gr)酶处理,探讨能否检测目的超螺旋结构的环状dna。通过将向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加15kb环状dna至成为0.5pm(5pg/μl)的扩增反应液(10μl)于30℃保温20小时来进行扩增反应。作为模板dna使用的15kb的环状dna使用使大肠杆菌基因组上的15kb区域和oric片段(0.4kb)连接环化,使用大肠杆菌而克隆化后纯化的。对于扩增反应后产物而用20ml的10mmtris-hcl(ph8.0)进行2小时透析,将其中0.5μl添加到含gr酶的反应缓冲液5μl中,于30℃20分钟或温育60分钟。作为gr酶,使用exoiii、dna聚合酶i、连接酶及促旋酶的组合,各自以20mu/μl、50nm、50nm、50nm的浓度添加。反应缓冲液使用表6中所示的组成。作为由gr酶的处理的阳性对照,使用作为引入切口的dna的phi×174rfii(neb公司)而进行缺口修复反应。缺口修复适合地进行,则切口被修复,可检测到超螺旋结构的环状dna。将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图35。在本实验中,由于高于最适于15kb的环状dna的扩增时间(3小时左右)的长时间反应,变得几乎无法观察到超螺旋产物。通过将此产物用gr酶处理,可检测到超螺旋结构的环状dna。这表明,可从扩增反应后的副产物,由缺口修复得到目的超螺旋结构的环状dna。【实施例14:长链环状dna的稳定化因子及使用其的扩增反应效率化】(1)长链dna的稳定化因子的探讨将长链环状dna用从表6除肌酸激酶和牛血清白蛋白的组成的反应缓冲液,于37℃温育,则观察到dna损伤被诱导,超螺旋结构的环状dna减少的样式。对于贡献于长链环状dna的稳定化的试剂而进行探讨。探讨的结果,得知葡萄糖、蔗糖、二甲基亚砜(dmso)、牛血清白蛋白(bsa)、二醇醚二胺四醋酸(egta)、浴铜灵二磺酸二钠(bda)、青霉胺、钛铁试剂(tiron、1,2-二羟基苯-3,5-磺酸酯)、二亚乙基三胺五醋酸(dtpa)、乙二胺四醋酸(edta)、dps蛋白质、(大肠杆菌来源)、金属硫蛋白质(人来源)对在反应缓冲液中的保温时的dna的稳定化显示效果。(2)由dna稳定化因子的环状dna扩增反应效率化通过将向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加200kb环状dna至成为0.5pm(67pg/μl)、加dtpa或tiron至成为0.05、0.1或0.3mm、加bda至成为0.1、0.3或1mm、加dps至成为0.3、1或3μm的扩增反应液(10μl)于30℃保温20小时进行扩增反应。将扩增后的反应液与实施例11同样地,于30℃1小时、稀释再保温之后,将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图36。得知(1)中发现的dna稳定化因子之中,在添加dtpa、tiron、bda、dps蛋白质及bsa的扩增反应物中,超螺旋结构的环状dna的产生量提升,有使环状dna扩增反应效率化的作用。【实施例15:使用乳液的长链环状dna的扩增反应】探讨在油包水滴型乳液内的环状dna的扩增反应。作为模板dna,使用实施例1中记载的200kb环状dna。调制向含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液加200kb环状dna至成为0.5pm(67pg/μl)的扩增反应液(5μl)。向此扩增反应液添加含表面活性剂(2%abilem90及0.05%triton-x100)的矿物油100μl,通过经60秒钟涡旋而混合。通过将此混合物于30℃保温3小时或18小时时间而进行扩增反应(乳液)。另外,为了比较,通过将上述的扩增反应液直接于30℃保温3小时或18小时来进行扩增反应(整体)。将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图37。在整体的系统中,见到低分子的副产物的生成的同时,变得无法观察到反应时间变长和目的超螺旋结构的dna。在乳液的系统中,低分子的副产物的生成被抑制的同时,对应于反应时间而目的超螺旋结构的dna的产生提升。【实施例16:由温度循环的扩增效率化】在环状dna的扩增反应中,由于越是低分子则复制越快速结束,作为副产物发生低分子的环状dna,则副产物的方快速地扩增。对于长链dna的扩增而言,由此现象而副产物的扩增变得显著,变得无法见到作为目的产物的长链dna的扩增是问题。为了有效扩增长链dna,有抑制低分子dna的过量扩增的必要。其中,发明人关注于对于含oric的环状dna的复制起始而言最适是30℃以上的温度,而延伸-分离反应在更低温也进行的点。尝试通过在环状dna的扩增反应中进行温度循环来具备复制起始的循环,抑制低分子dna的过量扩增。调制将含表6中所示的组成的反应缓冲液及表1中所示的组成的酶组的反应液根据实施例8于30℃预温育30分钟之后,加200kb环状dna(实施例1)至成为0.5pm(67pg/μl)的扩增反应液(10μl)。对于此扩增反应液而实施30个37℃、5分钟→16℃或24℃、30分钟的温度循环(2-步循环)。另外,作为用于比较的试样,将上述的扩增反应液于30℃保温21小时。将反应产物与实施例1同样地供于琼脂糖凝胶电泳而检测dna。结果示于图38。在以2-步循环反应时,低分子的副产物的产生被抑制的同时,目的超螺旋结构的dna的产生量增大。【产业上的利用可能性】由本发明可提供不使用大肠杆菌细胞或质粒载体而可简便并且指数扩增环状dna、特别是长链环状dna的方法。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1