用于治疗和预防动物的过敏性和/或炎症性疾病的取代吲唑的制作方法
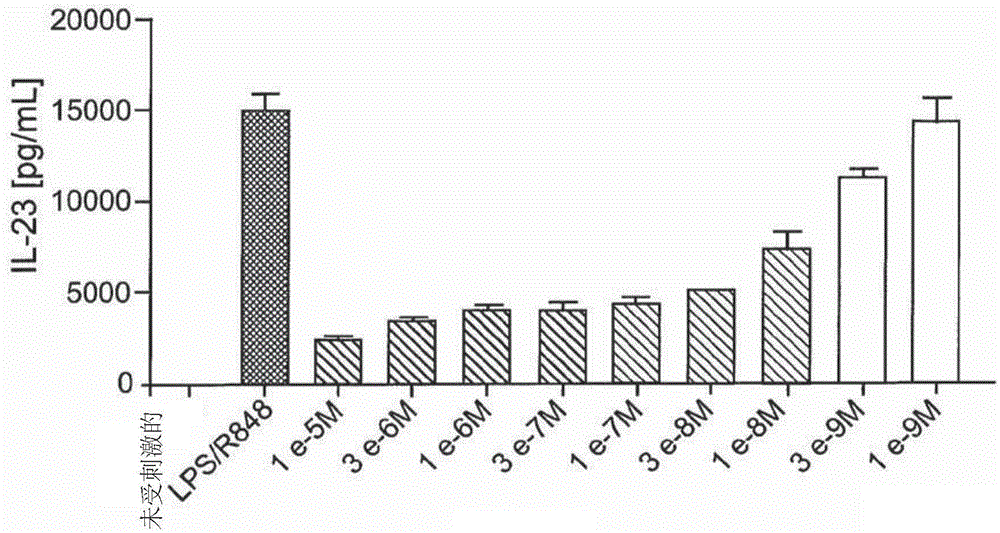
本发明涉及抑制白细胞介素-1受体相关激酶4(irak4)的通式(i)的新的取代吲唑的用途。人irak4(白细胞介素-1受体相关激酶4)在激活免疫系统中起关键作用。因此,该激酶是用于开发炎症抑制物质的重要的治疗靶分子。irak4由许多细胞表达,并介导以下受体的信号转导:toll样受体(tlr)(tlr3除外),以及由1l-1r(受体)、1l-18r、1l-33r和1l-36r组成的白细胞介素(1l)-1β家族受体(janeway和medzhitov,annu.rev.immunol.,2002;dinarello,annu.rev.immunol.,2009;flanneryandbowie,biochemicalpharmacology,2010)。irak4缺失的小鼠和irak4缺失的患者人细胞都不会对tlr(tlr3除外)和1l-1β家族的刺激起反应(suzuki,suzuki等人,nature,2002;davidson,currie等人,thejournalofimmunology,2006;ku,vonbernuth等人,jem,2007;kim,staschke等人,jem,2007)。tlr配体或1l-1β家族配体与各自受体的结合导致myd88[髓细胞分化初次应答基因(88)]募集并结合至受体。从而,myd88与irak4相互作用,导致形成活性复合物,所述活性复合物与激酶irak1或irak2相互作用并将其激活(kollewe,mackensen等人,journalofbiologicalchemistry,2004;precious等人,j.biol.chem.,2009)。由此,nf(核因子)-κb信号途径和mapk(促分裂原活化蛋白激酶)信号途径被激活(wang,deng等人,nature,2001)。nf-κb信号途径和mapk信号途径两者的激活产生与不同的免疫过程相关的过程。例如,存在增多的多种炎症信号分子和酶如细胞因子、趋化因子和cox-2(环氧化酶-2)的表达增加,以及增强的炎症相关基因如cox-2、il-6(白细胞介素-6)、il-8的mrna稳定性(holtmann,enninga等人,journalofbiologicalchemistry,2001;datta,novotny等人,thejournalofimmunology,2004)。此外,这些过程可伴随有某些细胞类型如单核细胞、巨噬细胞、树突细胞、t细胞和b细胞的增殖和分化(wan,chi等人,natimmunol,2006;mcgettrick和j.o'neill,britishjournalofhaematology,2007)。irak4在各种炎症性疾病的病理学中的核心作用已通过直接比较野生型(wt)小鼠与具有激酶灭活形式的irak4(irak4kdki)的基因修饰动物而示出。在多发性硬化、动脉粥样硬化、心肌梗塞和阿尔茨海默氏病的动物模型中,irak4kdki动物具有改善的临床表现(rekhter,staschke等人,biochemicalandbiophysicalresearchcommunication,2008;maekawa,mizue等人,circulation,2009;staschke,dong等人,thejournalofimmunology,2009;kim,febbraio等人,thejournalofimmunology,2011;cameron,tse等人,thejournalofneuroscience,2012)。此外,已发现,在动物模型中irak4的缺失通过改善抗病毒反应并同时减少全身炎症而防止病毒诱导的心肌炎(valaperti,nishii等人,circulation,2013)。还已表明irak4的表达与伏格特-小柳-原田综合症(vogt-koyanagi-haradasyndrome)的疾病活性相关(sun,yang等人,plosone,2014)。此外,已显示了irak4与通过浆细胞样树突细胞的免疫复合物介导的ifnα(干扰素-α)产生(系统性红斑狼疮(sle)发病中的关键过程)高度相关(chiang等人,thejournalofimmunology,2010)。此外,信号途径与肥胖相关(ahmad,r.,p.shihab等人,diabetology&metabolicsyndrome,2015)。除irak4在先天性免疫中的重要作用外,还存在这样的暗示:irak4影响th17t细胞(适应性免疫的成员)的分化。在irak4激酶活性缺失情况下,与wt小鼠相比,有较少的1l-17-生产t细胞(th17t细胞)产生。抑制irak4使得能够预防和/或治疗动脉粥样硬化、1型糖尿病、类风湿性关节炎、脊柱关节炎(尤其是牛皮癣性脊柱关节炎和别克捷列夫氏病(bekhterev'sdisease))、红斑狼疮、银屑病、白癜风、巨细胞动脉炎、慢性炎症性肠道疾病和病毒性疾病,例如hiv(人免疫缺陷病毒)、肝炎病毒(staschke等人,thejournalofimmunology,2009;marquez等人,annrheumdis,2014;zambrano-zaragoza等人,internationaljournalofinflammation,2014;wang等人,experimentalandtherapeuticmedicine,2015;ciccia等人,rheumatology,2015)。由于irak4在tlr(tlr3除外)和1l-1受体家族的myd88介导信号级联中的核心作用,抑制irak4可用于预防和/或治疗由所述受体介导的疾病。现有技术公开了许多irak4抑制剂(参见,例如annualreportsinmedicinalchemistry(2014),49,117–133)。us8293923和us20130274241公开了具有3-取代吲唑结构的irak4抑制剂。没有2-取代吲唑的记载。wo2013106254和wo2011153588公开了2,3-二取代吲唑衍生物。wo2007091107记载了2-取代吲唑衍生物用于治疗杜氏肌营养不良(duchennemusculardystrophy)。所公开的化合物不具有6-羟烷基取代基。wo2015091426记载了在2位被羧酰胺侧链取代的吲唑,如实施例64。实施例64wo2015104662公开了以下通式的2-取代吲唑:其中,r2为烷基或环烷基基团。在wo2015104662中明确记载了在2位具有甲基、2-甲氧基乙基和环戊基基团的2-取代吲唑(实施例1、4和76)。还由实施例117记载了在1位具有羟乙基取代基的吲唑衍生物。然而,没有记载在1位或2位具有3-羟基-3-甲基丁基取代基的吲唑衍生物。在wo2015104662中,通过通式宽泛地包括了在2位具有羟基取代的烷基基团的吲唑,但是没有明确地公开。在wo2015104662中,通式和r2取代基的定义没有包括在2位具有烷基基团且其中该烷基基团另外被甲基磺酰基取代的吲唑。除了吲唑上在1位和2位的上述取代形式外,wo2015104662还记载了在6位具有取代基、r1定义如下的吲唑:烷基、氰基、-nrarb或选自环烷基、芳基或杂环基的任选的取代基团,其中所述取代基独立地为烷基、烷氧基、卤素、羟基、羟烷基、氨基、氨基烷基、硝基、氰基、卤代烷基、卤代烷氧基、-ococh2-o-烷基、-op(o)(o-烷基)2或-ch2-op(o)(o-烷基)2。对于其中r1为烷基基团的吲唑化合物,有效的申请日为2015年1月7日(wo2015104662的国际申请日)。其要求优先权的印度申请146/che/2014和3018/che/2014并没有公开任何r1为烷基基团的吲唑化合物。因此,以下通式的吲唑化合物:其中r1为任选取代的烷基基团的吲唑化合物在2015年1月7日被第一次公开,因此是在本申请的优先权日之后。在wo2015104662中记载的6位取代基r1的实例为环丙基、环己基、氰基、3-氟苯基和饱和的杂环取代基。在wo2015104662中没有明确记载在6位具有羟基取代的烷基基团的吲唑。在2016年6月2日公布为wo2016083433的共同未决的专利申请pct/ep2015/077596中也记载了本发明中所使用的化合物。目前对动物的过敏性和/或炎症性疾病如过敏性皮肤病的治疗方案通常包括使用类固醇和环孢菌素—二者均有副作用。最近,janus-激酶(jak)抑制剂已被批准用于犬特应性皮炎(cad),其在症状上缓解瘙痒,然而,给药方案可能再次受到副作用的限制。使用疾病调节剂治疗cad且没有治疗相关的副作用,这仍然是未满足的医疗需求。本发明所解决的问题是为动物的炎症性和/或过敏性疾病提供更好的治疗方案。本发明的irak4抑制剂尤其适用于治疗和预防以过度反应的免疫系统为特征的动物炎症性疾病。这里应特别提及以下疾病:狗和猫的犬特应性皮炎、跳蚤过敏性皮炎;狗和猫的炎症性肠道疾病;狗、猫、马和牛的骨关节炎疼痛和炎症性疼痛;马的非感染性复发性气道疾病(又称慢性阻塞性肺病、肺气肿);马的昆虫过敏症(又称甜痒病、夏季湿疹);猫哮喘;牛呼吸道疾病;牛的乳腺炎和子宫内膜炎;以及猪呼吸道疾病。例如,特应性皮炎是宠物、特别是猫和狗中的一种常见疾病。作为一个特定实例,犬特应性皮炎(cad)是狗的最常见疾病之一。cad可从早期开始就影响患者,在其一生中反复发作。在lund等人1999年的一项研究中,研究调查了美国的52个私人诊所中的31,484只狗,cad的患病率为8.7%。cad是继跳蚤过敏性皮炎(fad)之后,犬瘙痒症的第二种最常见的原因。犬特应性皮炎可定义为“具有与ige相关、最常见针对环境过敏原的特征性临床特征的遗传易感性炎症和瘙痒性过敏性皮肤病”(halliwell,veterinaryimmunologyandimmunopathology,2006),如尘螨和花粉,由于尘螨几乎无处不在,并且花粉遍布室外空气,因此对于宠物来说,避免尘螨和花粉超乎想象地困难。犬特应性皮炎是一种复杂的多因素疾病,涉及免疫失调、过敏性过敏、皮肤屏障缺陷、微生物定植和环境因素。在所有的病例中,ige不是临床症状发展的先决条件,并且被称为类特应性皮炎的单独的临床实体而被定义为“具有与在犬特应性皮炎中观察到的那些临床特征相同的临床特征的炎症性和瘙痒性皮肤病,在犬特应性皮炎中,对环境或其他过敏原的ige应答未从文献中找到”(nuttall等人,vet.record,2013)。犬特应性皮炎最常见的症状包括瘙痒、过度抓挠、在地毯上摩擦、掉毛、有臭味的油腻或片状皮肤、爪子以及腹股沟和腋窝等区域的过度咀嚼。随着时间推移,被抓挠的皮肤可能产生可能会被感染的热点—刺痛的(raw)、发炎的区域。目前,治疗急性发作(flares)的特应性皮炎(ad)应包括寻找并且然后消除发作的原因,用温和的沐浴液沐浴,并且采取包括外用(topical)和/或口服糖皮质激素或奥拉替尼(oclacitinib)的干预措施控制瘙痒和皮肤病变。对于慢性cad,管理的第一步是识别并避免发作因素,以及确保有足够皮肤和表面卫生护理;这可能包括更频繁的洗浴和可能增加必需的脂肪酸摄入。当前在减轻慢性瘙痒和皮肤损伤方面最有效的药物为外用和口服的糖皮质激素、口服的环孢菌素、口服的奥拉替尼,以及在可获得的情况下,可注射的重组干扰素。过敏原特异性免疫治疗和主动间歇性外用的糖皮质激素应用是唯一可能预防或延缓ad发作的复发的干预措施(olivry等人,bmcveterinaryresearch,2015)。作为另一个具体实施例,跳蚤过敏性皮炎(fad)或跳蚤叮咬过敏是家犬的最常见的皮肤病(scott等人,in:mullerandkirk’ssmallanimaldermatology,2001),由迄今为止狗和猫上最常见的跳蚤:猫栉首蚤(ctenocephalidesfelis)(beresford-jones,jsmallanimalpractice,1981;chesney,veterinaryrecord,1995)引起。猫也会产生fad,这是猫粟粒性皮炎的主要病因之一。fad在夏季最为普遍,然而在温暖的气候中跳蚤感染可能持续整年。在北温带区域,宠物及其跳蚤与人类住所的密切联系创造了条件,使得产生全年性问题。极端温度和低湿度往往会抑制跳蚤的发展。在进食时,跳蚤注射含有多种组胺类化合物、酶、多肽和大小跨度范围宽泛(40–60kd)的氨基酸的唾液,从而诱导i型、iv型和嗜碱性粒细胞过敏症。间歇性暴露于跳蚤叮咬的跳蚤-幼犬产生立即(15min)反应或延迟(24-48小时)反应,或二者都有,并产生可检测水平的循环ige和igg抗跳蚤抗体。连续暴露于跳蚤叮咬的狗具有低水平的这些循环抗体,并且也不产生皮肤反应,或者稍后产生并且至相当低的程度。这可能表明连续暴露于跳蚤叮咬的狗中自然地产生了免疫耐受性。虽然对猫的fad病理生理学知之甚少,但类似的机理可能存在。猫蚤(猫栉首蚤)对动物和人造成严重刺激,并导致跳蚤过敏性皮炎。典型的症状为:瘙痒、皮肤炎症和皮肤病变(红斑、鳞屑、丘疹、结痂和苔藓化)。这些损害最常见于背部和尾根部。随着病情发展,可能会出现掉毛、断毛、渗出(oozing)或痂疮溃疡(crustysores)、疙瘩肿块(pimplybumps)、全身发红和皮肤发炎。溃疡可能非常疼痛。在严重的情况下,皮肤会变厚和变暗,主要在狗的背部和尾根部区域。由于严重的瘙痒,狗本身会造成自残的伤害。一般来说,预防和治疗跳蚤感染是选择的治疗方案。使用最常见的新烟碱类如吡虫啉(imidacloprid),或γ-氨基丁酸(gaba)-门控氯通道阻断剂如氟虫腈(fipronil)。如果皮肤过敏性皮炎症状不消除,则使用cad下所提及的目前的治疗,如外用和口服糖皮质激素、口服环孢菌素、口服奥拉替尼。本发明提供通式(i)的化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病在式(i)中:r1为c1-c6-烷基,其中c1-c6-烷基基团未被取代,或者被以下基团单取代或相同或不同地多取代卤素、羟基、未取代的或者单或多-卤素-取代的c3-c6-环烷基,或r6、r7so2、r7so或r8o基团,或选自以下的基团:其中*代表基团至分子其余部分的键合位点;r2和r3总是具有相同的定义,并均为氢或c1-c6-烷基;r4为卤素、氰基、未取代的c1-c6-烷基或者单取代或相同或不同地多取代的c1-c6-烷基、或未取代的c3-c6-环烷基或者单取代或相同或不同地多取代的c3-c6-环烷基,取代基选自卤素和羟基;r5为氢、卤素、或未取代的或者单或多-卤素-取代的c1-c6-烷基;r6为具有4至6个环原子的未取代或者单-或二-甲基-取代的单环饱和杂环,其含有选自o、s、so和so2的杂原子或杂基;r7为c1-c6-烷基,其中c1-c6-烷基基团未被取代,或者被卤素、羟基或c3-c6-环烷基单取代或者相同或不同地多取代,或r7为c3-c6-环烷基;r8为c1-c6-烷基,其中c1-c6-烷基基团未被取代,或者被卤素单取代或者相同或不同地多取代。在下文描述的本发明的合成中间体和工作实施例的所述用途的情况下,以相应的碱或酸的盐的形式指定的任何化合物,通常是通过相应的制备和/或纯化方法获得的未知确切化学计量组成的盐。因此,对于这样的盐,除非更详细地说明,否则另外的名称和结构式如“盐酸盐”、“三氟乙酸盐”、“钠盐”或“xhci”、“xcf3cooh”、“xna+”不应当以化学计量意义理解,而仅是对其中存在的盐形成组分的说明性符号。这也相应适用于通过所述制备和/或纯化方法获得的化学计量组成未知(如果其为所定义类型)的溶剂合物如水合物形式的合成中间体或工作实施例或其盐的情况。本发明化合物为式(i)的化合物及其盐、溶剂合物和盐的溶剂合物,由式(i)涵盖且为下述结构式的化合物及其盐、溶剂合物和盐的溶剂合物,以及由式(i)涵盖并在下文中作为实施方案提及的化合物及其盐、溶剂合物和盐的溶剂合物,即使由式(i)涵盖并在下文中提及的化合物并不是盐、溶剂合物和盐的溶剂合物。在本发明上下文中,优选的盐为本发明化合物的生理上可接受的盐。然而,本公开还涵盖本身不适用于药物应用但可用于例如本发明化合物的分离或纯化的盐。本发明化合物的生理上可接受的盐包括无机酸、羧酸和磺酸的酸加成盐,例如如下酸的盐:盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、三氟乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸的盐。本发明化合物的生理上可接受的盐还包括常规碱的盐,例如并且优选碱金属盐(例如钠盐和钾盐)、碱土金属盐(例如钙盐和镁盐),以及衍生自氨或具有1至16个碳原子的有机胺的铵盐,例如并且优选乙胺、二乙胺、三乙胺、乙基二异丙基胺、单乙醇胺、二乙醇胺、三乙醇胺、二环己基胺、二甲氨基乙醇、普鲁卡因、二苄基胺、n-甲基吗啉、精氨酸、赖氨酸、乙二胺和n-甲基哌啶。在本发明上下文中,溶剂合物描述为通过与溶剂分子配位形成固态或液态络合物的本发明化合物的那些形式。水合物是与水配位的特殊形式的溶剂合物。本发明化合物,根据其结构,可以以不同的立体异构形式存在,即以构型异构体或者(如果合适)构象异构体(对映异构体和/或非对映异构体,包括阻转异构体)的形式存在。因此,本发明包括对映异构体和非对映异构体及其各自的混合物的用途。可以以已知方式从对映异构体和/或非对映异构体的这些混合物中分离出立体异构均一的成分;为此目的优选使用色谱法,尤其是非手性相或手性相上的hplc色谱法。如果本发明化合物可以以互变异构形式存在,则本发明包括所有的互变异构形式的用途。本发明还包括本发明化合物的所有合适的同位素变体的用途。在本文中,本发明化合物的同位素变体应理解为意指这样的化合物:其中本发明化合物中至少一个原子已被替换为原子序数相同,但原子质量与在自然界中通常或主要存在的原子质量不同的另一原子。可并入本发明化合物的同位素的实例为氢、碳、氮、氧、磷、硫、氟、氯、溴和碘的那些同位素,例如2h(氘)、3h(氚)、13c、14c、15n、17o、18o、32p、33p、33s、34s、35s、36s、18f、36cl、82br、123i、124i、129i和131i。本发明化合物的特别的同位素变体,例如特别是其中已并入一种或多种放射性同位素的那些变体,可有利于考察在体内的作用机理或活性成分分布;由于相对容易的可制备性和可检测性,特别是用3h或14c同位素标记的化合物适合于此目的。此外,并入同位素(例如氘)由于化合物的更高的代谢稳定性可能产生特别的治疗益处,例如在体内的半衰期延长或所需的活性剂量减少;因此,在某些情况下,对本发明化合物的这样的修饰还可构成本发明用途的优选实施方案。本发明化合物的同位素变体可通过本领域技术人员已知的方法制备,例如通过在下文中进一步说明的方法和在工作实施例中记载的方法,通过使用各自试剂和/或起始化合物的相应同位素修饰来制备。本发明还提供本发明化合物的所有可能的晶型和多晶型的用途,其中所述多晶型物可作为单一多晶型物或所有浓度范围内的诸多多晶型物的混合物存在。此外,本发明还包括本发明化合物的前药的用途。在本上下文中,术语“前药”指的是本身可以是生物活性或无活性的,但在体内停留期间被(例如代谢或水解地)转化为本发明化合物的化合物。在本发明上下文中,除非另有说明,取代基具有以下含义:烷基在本发明上下文中表示具有指定的特定数目碳原子的直链或支链的烷基基团。实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、1-甲基丙基、2-甲基丙基、叔丁基、正戊基、1-乙基丙基、1-甲基丁基、2-甲基丁基、3-甲基丁基、2,2-二甲基丙基、正己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1-乙基丁基和2-乙基丁基。优选甲基、乙基、正丙基、正丁基、2-甲基丁基、3-甲基丁基和2,2-二甲基丙基。环烷基在本发明上下文中为在每种情况下具有指定数目的碳原子的单环饱和烷基基团。优选的实例包括环丙基、环丁基、环戊基和环己基。烷氧基在本发明上下文中表示具有指定的特定数目碳原子的直链或支链烷氧基基团。优选1至6个碳原子。实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、1-甲基丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基、1-乙基丙氧基、1-甲基丁氧基、2-甲基丁氧基、3-甲基丁氧基和正己氧基。特别优选具有1至4个碳原子的直链或支链的烷氧基基团。作为优选的提及的实例为甲氧基、乙氧基、正丙氧基、1-甲基丙氧基、正丁氧基和异丁氧基。卤素在本发明上下文中为氟、氯和溴。优选氟。羟基在本发明上下文中为oh。单环饱和杂环为具有4至6个环原子并且含有选自o、s、so和so2的杂原子或杂基的单环饱和杂环。优选具有选自o、so和so2的杂原子或杂基的杂环。实例包括:氧杂环丁烷、四氢呋喃、四氢-2h-吡喃-4-基、1,1-二氧四氢-2h-噻喃-3-基、1,1-二氧四氢-2h-噻喃-2-基、1,1-二氧四氢-2h-噻喃-4-基、1,1-二氧四氢噻吩-3-基、1,1-二氧四氢噻吩-2-基、1,1-二氧硫杂环丁-2-基或1,1-二氧硫杂环丁-3-基。在本文中特别优选氧杂环丁烷和四氢呋喃。非常特别优选氧杂环丁烷-3-基。键上的符号*表示分子中的键合位点。除非另有说明,当本发明化合物中的基团被取代时,所述基团可被单取代或多取代。在本发明上下文中,出现多于一次的所有基团均彼此独立地定义。优选被一个、两个或三个相同或不同的取代基取代。r1的一个优选实施方案为被1、2或3个氟原子取代的c2-c6-烷基基团。特别优选2,2,2-三氟乙基、3,3,3-三氟丙基和4,4,4-三氟丁基。非常特别优选4,4,4-三氟丁基基团。r1的另一优选实施方案为被一个或两个羟基基团或一个c1-c3-烷氧基或三氟取代的c1-c3-烷氧基取代的c2-c6-烷基基团。特别优选被羟基或c1-c3-烷氧基或三氟甲氧基或2,2,2-三氟乙氧基取代的c2-c5-烷基基团。非常特别优选3-羟基-3-甲基丁基、3-甲氧基丙基、3-羟丙基、3-三氟甲氧基丙基、2-甲氧基乙基或2-羟乙基。尤其优选3-羟基-3-甲基丁基基团。另外优选地,r1为被c1-c6-烷基-so2基团取代的c2-c6-烷基基团。特别优选甲基-so2-取代的c2-c4-烷基。尤其优选r1为2-(甲基磺酰基)乙基或3-(甲基磺酰基)丙基。在后一组中,特别优选2-(甲基磺酰基)乙基。另外,优选地,r1为被以下基团取代的c1-c3-烷基基团:氧杂环丁烷基、四氢呋喃基、四氢-2h-吡喃-4-基、1,1-二氧四氢-2h-噻喃-3-基、1,1-二氧四氢-2h-噻喃-2-基、1,1-二氧四氢-2h-噻喃-4-基、1,1-二氧四氢噻吩-3-基、1,1-二氧四氢噻吩-2-基、1,1-二氧硫杂环丁-2-基或1,1-二氧硫杂环丁-3-基。特别优选被氧杂环丁烷基团取代的c1-c3-烷基基团。尤其优选r1为氧杂环丁烷-3-基甲基基团。对于r2和r3,总是具有相同的定义,优选氢或甲基。特别优选甲基。对于r4,优选未取代或者单或多-卤素-取代的c1-c3-烷基基团,或被一个羟基基团取代的c1-c3-烷基基团,或被一个羟基基团和三个氟原子取代的c1-c3-烷基基团。对于r4,特别优选以下基团:甲基、乙基、三氟-c1-c3-烷基、二氟-c1-c3-烷基、羟甲基、1-羟乙基、2-羟基丙-2-基和2,2,2-三氟-1-羟乙基。对于r4,特别优选甲基、三氟甲基和二氟甲基基团。本文中特别优选三氟甲基基团。r5的优选实施方案为氢、氟、氯或c1-c3-烷基。更优选地,r5为氢、氟或甲基。最优选地,r5为氢或氟。还特别优选r4为甲基或三氟甲基并且r5为氟的化合物。非常特别优选r4为甲基并且r5为氟的化合物,其中r5在r4的邻位。对于r6,优选的实施方案包括氧杂环丁烷基、四氢呋喃基、四氢-2h-吡喃-4-基、1,1-二氧四氢-2h-噻喃-3-基、1,1-二氧四氢-2h-噻喃-2-基、1,1-二氧四氢-2h-噻喃-4-基、1,1-二氧四氢噻吩-3-基、1,1-二氧四氢噻吩-2-基、1,1-二氧硫杂环丁-2-基或1,1-二氧硫杂环丁-3-基。本文中特别优选氧杂环丁烷基。非常特别优选氧杂环丁烷-3-基。r7仅与官能团-so2-和-so-连接,即为r7-取代的-so2-或so基团。对此,r7优选为c1-c4-烷基,其中c1-c4-烷基基团未被取代或被羟基或环丙基单取代或被三个氟原子取代。另外,优选r7为环丙基基团。特别优选r7为甲基、乙基或羟乙基。非常特别优选r7为甲基。这意味着,在被r7so2-或r7so-取代的c1-c6-烷基基团的情况下,就r1而言,优选被c1-c6-烷基-so2或c1-c6-烷基-so取代的c1-c6-烷基。对于r1,在本文中尤其优选甲基磺酰基乙基和甲基磺酰基丙基。在本文中非常特别优选甲基磺酰基乙基。对于r8,优选未被取代的c1-c4-烷基基团或三氟取代的c1-c4-烷基基团。特别优选甲基、乙基、三氟甲基或2,2,2-三氟乙基。非常特别优选甲基、三氟甲基或2,2,2-三氟乙基。如前所述,细胞内酶白细胞介素-1受体相关激酶4(irak4)在由细胞因子和tlr配体激活的受体的信号传导途径中起着不可或缺的作用,所述细胞因子和tlr配体涉及炎症过程。除炎症外,irak4还参与过敏过程的信号传导。这样的过敏过程在过敏性皮肤病如特应性皮炎的发病机理中起重要作用。例如,最近添加到还包括il-18和il-1的il-1细胞因子家族中的il-33,键合至并激活il-33受体(il-33r),所述il-33受体(il-33r)又与myd88、irak4和trf6相关(schmitz等人,immunity,2005)。irak4是该信号传导途径的必要组分。il-33r在t辅助细胞2型(th2)细胞、肥大细胞和嗜酸性粒细胞中强烈表达。il-33激活这些细胞并促进th2的免疫应答(schmitz等人,immunity,2005)。这些细胞类型都参与了特应性皮炎的发病机理。血清中的il-33含量与人特应性皮炎的严重程度相关,且用外用类固醇&钙调神经酶抑制剂治疗时会降低(tamagawa-mineoka等人,jamericanacademydermatology,2014)。在急性犬特应性皮炎的模型中,已显示il-33基因在皮肤损伤中显著上调(schamber等人,g3(bethesda),2014;olivry等人,journalofinvestigativedermatology,2016)。此外,il-1细胞因子家族的第二个成员,il-18与特应性皮炎相关。血清il-18水平随着儿童特应性皮炎的严重程度而增加(sohn等人,allergyandasthmaproceedings,2004)。在急性犬特应性皮炎的模型中,已显示在皮肤损伤中il-18基因显著上调(schamber等人,g3(bethesda),2014;olivry等人,journalofinvestigativedermatology,2016)。此外,特应性皮炎类炎症&瘙痒由il-18的过度释放引起,并通过小鼠il-1加速(konishi等人,proceedingsofthenationalacademyofsciences,2002)。此外,已显示irak-4是il-18信号级联的必需组分(suzuki等人,jimmunology,2003)。类似地,irak4对于il-1和tlr配体的信号传导至关重要(suzuki等人,nature,2002)。已知tlr拮抗剂可引起瘙痒(liu等人,neurosciencebulletin,2012)—特应性皮炎的重要症状,并将抗il-1疗法标签外地用于治疗特应性皮炎。此外,irak4的多晶型与过敏性疾病如哮喘和慢性鼻窦炎中的总ige升高相关(tewfik等人,allergy,2009)。在特应性皮炎中,ige水平也升高。因此,由于irak4是由许多细胞因子、tlr配体激活的信号途径的关键部分,并且irak4具有与ige水平增加相关的多晶型,因此抑制irak4是治疗过敏性皮肤病如特应性皮炎的重要治疗策略。此外,在宠物(尤其是狗和猫)中,特应性皮炎和跳蚤过敏性皮炎都是适当的适应症,因为两种疾病都包括涉及ige抗体、th2细胞、肥大细胞和嗜酸性粒细胞的i型过敏症。此外,fad可包括其中涉及il-1和il-18的iv型过敏症。本发明化合物起irak4激酶抑制剂的作用,因此在治疗和/或预防动物的过敏性和/或炎症性疾病中具有不可预见的有用的药理学活性谱。优选式(i)的化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,在式(i)中r1为c1-c6-烷基,其中c1-c6-烷基基团未被取代或者被氟、羟基,或r6、r7so2、r7so或r8o基团单取代或相同或不同地多取代;r2和r3总是具有相同的定义,并且都为氢或c1-c3-烷基;r4为卤素、氰基或c1-c3-烷基,其中c1-c3-烷基基团未被取代或者被卤素或羟基单取代或相同或不同地多取代;r5为氢、氟、氯或c1-c3-烷基;r6为氧杂环丁烷基或四氢呋喃基;r7为c1-c4-烷基,其中c1-c4-烷基基团未被取代,或被羟基或环丙基单取代,或被三个氟原子取代;r8为未被取代的c1-c4-烷基或三氟取代的c1-c4-烷基。另外优选式(i)的化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,在式(i)中r1为c2-c6-烷基,其中c2-c6-烷基未被取代,或c2-c6-烷基被单-、二-或三-氟取代,或c2-c6-烷基被羟基、r6、r7so2或r8o单取代,或其中r1为氧杂环丁烷基-取代的c1-c3-烷基;r2和r3总是具有相同的定义,并且都为氢或甲基;r4为未取代的或单-或多-卤素-取代的c1-c3-烷基,或被一个羟基取代的c1-c3-烷基基团,或被一个羟基和三个氟原子取代的c1-c3-烷基基团;r5为氢、氟或c1-c3-烷基;r7为c1-c3-烷基;r8为c1-c4-烷基,其中c1-c4-烷基基团为未取代的或单-、二-或三-氟取代的。还特别优选通式(i)的化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,在通式(i)中r1为被羟基,或c1-c3-烷氧基,或三氟甲氧基,或2,2,2-三氟乙氧基,或三氟甲基取代的c2-c5-烷基基团,或为甲基-so2-取代的c2-c4-烷基基团,或为氧杂环丁烷-3-基取代的c1-c2-烷基基团;r2和r3总是具有相同的定义,并且都为氢或甲基;r4为甲基、乙基、三氟-c1-c3-烷基、二氟-c1-c3-烷基、羟甲基、1-羟乙基、2-羟基丙-2-基和2,2,2-三氟-1-羟乙基,以及r5为氢、氟或甲基。非常特别优选下述化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,其中r1为4,4,4-三氟丁基、3-羟基-3-甲基丁基、3-羟丁基、3-甲氧基丙基、3-羟丙基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-三氟甲氧基丙基、2-甲氧基乙基、2-羟乙基、2-(甲基磺酰基)乙基或3-(甲基磺酰基)丙基;r2和r3都为甲基或氢,和r4为二氟甲基、三氟甲基或甲基,和r5为氢或氟。还非常特别优选下述化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,其中r1为3-羟基-3-甲基丁基、3-羟丁基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-(甲基磺酰基)丙基或2-(甲基磺酰基)乙基;r2和r3都为甲基;r4为二氟甲基或三氟甲基;和r5为氢。另外还特别优选下述化合物,及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病,其中r1为3-羟基-3-甲基丁基、3-羟丁基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-(甲基磺酰基)丙基或2-(甲基磺酰基)乙基;r2和r3都为甲基;r4为甲基,和r5为氟,其中r5在r4的邻位。本发明特别提供以下化合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病:1)n-[6-(2-羟基丙-2-基)-2-(2-甲氧基乙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺2)n-[6-(羟甲基)-2-(2-甲氧基乙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺3)n-[6-(2-羟基丙-2-基)-2-(3-甲氧基丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺4)n-[6-(羟甲基)-2-(3-甲氧基丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺5)n-[2-(2-羟乙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺6)n-[6-(2-羟基丙-2-基)-2-(3-羟丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺7)n-[2-(2-羟乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺8)n-[6-(2-羟基丙-2-基)-2-(氧杂环丁烷-3-基甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺9)n-[6-(羟甲基)-2-(氧杂环丁烷-3-基甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺10)n-{6-(2-羟基丙-2-基)-2-[3-(甲基磺酰基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺11)n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺12)n-{6-(2-羟基丙-2-基)-2-[2-(甲基磺酰基)乙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺13)6-(二氟甲基)-n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]吡啶-2-羧酰胺14)6-(二氟甲基)-n-{6-(2-羟基丙-2-基)-2-[2-(甲基磺酰基)乙基]-2h-吲唑-5-基}吡啶-2-羧酰胺15)6-(二氟甲基)-n-[6-(2-羟基丙-2-基)-2-(3-羟丙基)-2h-吲唑-5-基]吡啶-2-羧酰胺16)n-[6-(2-羟基丙-2-基)-2-(4,4,4-三氟丁基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺17)n-{6-(2-羟基丙-2-基)-2-[3-(三氟甲氧基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺18)n-{6-(2-羟基丙-2-基)-2-[3-(2,2,2-三氟乙氧基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺19)5-氟-n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-甲基吡啶-2-羧酰胺20)n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-甲基吡啶-2-羧酰胺21)6-(2-羟基丙-2-基)-n-[6-(2-羟基丙-2-基)-2-(4,4,4-三氟丁基)-2h-吲唑-5-基]吡啶-2-羧酰胺22)n-{2-[2-(1-羟基环丙基)乙基]-6-(2-羟基丙-2-基)-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺。本发明还提供通式(iii)的化合物及其非对映异构体、对映异构体、代谢物、盐、溶剂合物或盐的溶剂合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病在通式(iii)中r1为4,4,4-三氟丁基、3-羟基-3-甲基丁基、3-甲氧基丙基、3-羟丙基、3-羟丁基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-三氟甲氧基丙基、2-甲氧基乙基、2-羟乙基、2-(甲基磺酰基)乙基、3-(甲基磺酰基)丙基或2-(1-羟基环丙基)乙基;r4为二氟甲基、三氟甲基或甲基;以及r5为氢或氟。尤其优选以下通式(iii)的化合物,其用于治疗和/或预防动物的过敏性和/或炎症性疾病:5-{[(5-氟-6-甲基吡啶-2-基)羰基]氨基}-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯和2-(3-羟基-3-甲基丁基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯。通式(iii)的化合物适于制备一部分的通式(i)的化合物。此外,通式(iii)的化合物为白细胞介素-1受体相关激酶-4(irak4)的抑制剂。通式(iii)的化合物可由通式(ii)的化合物制备,通过使(ii)与适当取代的烷基卤化物或4-甲基苯磺酸烷基酯在碳酸钾存在下反应,其中r1为4,4,4-三氟丁基、3-羟基-3-甲基丁基、3-甲氧基丙基、3-羟丙基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-三氟甲氧基丙基、2-甲氧基乙基、2-羟乙基、2-(甲基磺酰基)乙基、3-(甲基磺酰基)丙基或2-(1-羟基环丙基)乙基;r4为二氟甲基、三氟甲基或甲基;和r5为氢或氟。此外,通式(i)的化合物可由式(iii)的化合物通过与甲基溴化镁的格氏反应而制备其中r1为4,4,4-三氟丁基、3-羟基-3-甲基丁基、3-羟丁基、3-甲氧基丙基、3-羟丙基、3-羟基-2-甲基丙基、3-羟基-2,2-二甲基丙基、3-三氟甲氧基丙基、2-甲氧基乙基、2-羟乙基、3-(甲基磺酰基)丙基、2-(1-羟基环丙基)乙基;r2和r3为甲基;r4为二氟甲基、三氟甲基或甲基;以及r5为氢或氟。本发明化合物起irak4激酶抑制剂的作用,并且具有不可预见的有用的药理学活性谱。进一步优选将式(i)的化合物,或上文特别提及的化合物用于治疗和/或预防家畜、特别是猫和狗、且更特别是狗的过敏性和/或炎症性疾病。在本上下文中,术语“家畜”包括例如哺乳动物,如仓鼠、豚鼠、大鼠、小鼠、龙猫、雪貂,或特别是狗、猫;笼养鸟;爬行动物;两栖动物或观赏鱼。另外优选将式(i)的化合物,或上文特别提及的化合物用于治疗和/或预防家畜的过敏性皮炎,特别是犬和猫过敏性皮炎,且更特别是犬过敏性皮炎。另外优选将式(i)的化合物,或上文特别提及的化合物用于治疗和/或预防农畜,特别是绵羊、山羊、马、牛和猪,且更特别是牛和猪的过敏性和/或炎症性疾病。在本上下文中,术语“农畜”包括例如哺乳动物,如马、绵羊、山羊、水牛、驯鹿、小鹿或更特别是牛或猪。另外优选将式(i)的化合物,或上文特别提及的化合物用在用于治疗和/或预防动物以下疾病的方法中:特应性皮炎、跳蚤过敏性皮炎、炎症性肠道疾病、骨关节炎疼痛和炎症性疼痛、非感染性复发性气道疾病、昆虫过敏症、哮喘、呼吸系统疾病、乳腺炎和子宫内膜炎,特别是特应性皮炎和跳蚤过敏性皮炎。特别优选将式(i)的化合物,或上文特别提及的化合物,用在用于治疗和/或预防以下疾病的方法中:狗或猫的犬特应性皮炎、跳蚤过敏性皮炎;狗或猫的炎症性肠道疾病;狗、猫、马或牛的骨关节炎疼痛和炎症性疼痛;马的非传染性复发性气道疾病;马的昆虫过敏症;猫哮喘;牛呼吸道疾病;牛乳腺炎;牛子宫内膜炎和猪呼吸道疾病。非常特别优选将式(i)的化合物或上文特别提及的化合物用在用于治疗和/或预防狗或猫、更特别地狗的犬特应性皮炎和跳蚤过敏性皮炎的方法中。另外非常特别优选将式(i)的化合物或上文特别提及的化合物用在用于治疗和/或预防牛骨关节炎疼痛和炎症性疼痛、牛呼吸道疾病、牛乳腺炎、牛子宫内膜炎和猪呼吸道疾病的方法中。关于式(iii)的化合物,另外优选将式(iii)的化合物用于治疗和/或预防家畜,特别是猫和狗,且更特别是狗的过敏性和/或炎症性疾病。另外优选将式(iii)的化合物用于治疗和/或预防家畜的过敏性皮炎,特别是犬和猫过敏性皮炎,且更特别是犬过敏性皮炎。另外优选将式(iii)的化合物用于治疗和/或预防农畜,特别是绵羊、山羊、马、牛和猪,且更特别是牛和猪的过敏性和/或炎症性疾病。另外优选将式(iii)的化合物用在用于治疗和/或预防动物的以下疾病的方法中:特应性皮炎、跳蚤过敏性皮炎、炎症性肠道疾病、骨关节炎疼痛和炎症性疼痛、非感染性复发性气道疾病、昆虫过敏症、哮喘、呼吸道疾病、乳腺炎和子宫内膜炎,特别是特应性皮炎和跳蚤过敏性皮炎。特别优选将式(iii)的化合物用在用于治疗和/或预防以下疾病的方法中:狗或猫的犬特应性皮炎、跳蚤过敏性皮炎;狗或猫的炎症性肠道疾病;狗、猫、马或牛的骨关节炎疼痛和炎症性疼痛;马的非传染性复发性气道疾病;马的昆虫过敏症;猫哮喘;牛呼吸道疾病;牛乳腺炎;牛子宫内膜炎和猪呼吸道疾病。非常特别优选将式(iii)的化合物用在用于治疗和/或预防狗或猫,更特别地狗的犬特应性皮炎和跳蚤过敏性皮炎的方法中。另外非常特别优选将式(iii)的化合物用在用于治疗和/或预防牛骨关节炎疼痛和炎症性疼痛、牛呼吸道疾病、牛乳腺炎、牛子宫内膜炎和猪呼吸道疾病的方法中。例如,化合物实施例11、12、13、19(如下所示)已在下文中详细记载的体外irak4tr-fret试验中使用重组犬irak4酶进行了评估。已经计算了各化合物对于抑制犬irak4的ic50值。已确认实施例化合物(11、12、13、19)可用于治疗动物、特别是狗和猫的过敏性皮肤病,例如特应性皮炎和跳蚤过敏性皮炎。实施例化合物11、12、13、19分别是ic50值分别为1.7、9.2、2.2、7.6nm的犬irak4的有效抑制剂。对于抑制人irak4,这些实施例化合物中的每一种的ic50值类似于所计算的ic50值。作为另一个实施例,还对实施例化合物12进行了体外试验评估,以确定化合物对脂多糖(lps)诱导的由犬外周血单核细胞(pbmc)细胞因子生成的功效。实施例化合物12通过lps诱导的犬pbmc以浓度相关方式而抑制促炎细胞因子肿瘤坏死因子α(tnfα)的生成。pbmc包括细胞类型如树突细胞、t淋巴细胞和b淋巴细胞,以及单核细胞,其每一种都与特应性皮炎相关,且在特应性皮炎患者中tnfα升高(sumimoto等人,archivesofdiseaseinchildhood,1992)。该实施例还由图7说明。因此,本发明化合物显示了犬pbmc对重组犬irak4酶和细胞因子生成的抑制,表明这些化合物实施例在犬特应性皮炎和跳蚤过敏性皮炎中的潜在的治疗益处。此外,在室内尘螨模型中,在进一步的研究中对实施例化合物12进行了体内评估,以确定化合物在治疗与犬过敏性皮炎,特别是犬特应性皮炎(cad)相关的临床症状中的功效。实施例化合物12显著减轻了cad类皮肤水肿和红斑的临床症状。该实施例还由图11和图12说明。因此,本发明化合物显示出犬过敏性皮炎的特征性临床症状的减轻,从而表明这样的化合物实施例在犬过敏性皮炎、特别是犬特应性皮炎(cad)中的治疗益处。此外,已在犬跳蚤过敏性皮炎(cad)的体内模型中对实施例化合物12进行了评估,以确定化合物的抗瘙痒功效。随后用实施例化合物12治疗可显著减轻了与过敏性疾病如跳蚤过敏性皮炎相关的瘙痒。该实施例还由图13说明。因此,本发明化合物显示了过敏性皮炎如皮肤炎和瘙痒相关的特征性临床症状的减轻,因而表明了这样的化合物实施例在犬过敏性皮炎、特别是跳蚤过敏性皮炎(fad)和犬特应性皮炎(cad)中的治疗益处。在本上下文中,术语“犬过敏性皮炎”特别包括犬特应性皮炎(cad)和跳蚤过敏性皮炎(fad)。作为另一个实施例,还已对实施例化合物12进行离体(exvivo)试验评估,以确定化合物对脂多糖(lps)诱导的由牛外周血单核细胞(pbmc)细胞因子生成的功效。实施例化合物12通过lps诱导的犬pbmc,以浓度相关的方式抑制促炎细胞因子肿瘤坏死因子α(tnfα)的生成。pbmc包括细胞类型如树突细胞、t淋巴细胞和b淋巴细胞,以及单核细胞,其每一种都与具有过度促炎免疫应答的炎症性疾病和感染性疾病如呼吸道疾病(sterner-kock,haider等人,tropicalanimalhealthandproduction,2016)、肠道疾病(pan,rostagnio等人,veterinaryimmunologyandimmunopathology,2015)和乳腺炎(zheng,xu等人,freeradicalbiologyandmedicine,2016)相关,在这些疾病的患者中tnfα升高。该实施例还由图8和9说明。因此,本发明化合物显示了牛pbmc对细胞因子生成的抑制,表明了这样的化合物实施例在炎症性疾病和/或感染性疾病如呼吸道疾病、肠道疾病和乳腺炎中的可能的治疗益处。作为另一个实施例,还已对实施例化合物12进行了离体试验评估,以确定化合物对脂多糖(lps)诱导的由猪外周血单核细胞(pbmc)生成细胞因子的功效。实施例化合物12通过lps诱导的猪pbmc抑制促炎细胞因子肿瘤坏死因子α(tnfα)的生成。pbmc包括细胞类型如树突细胞、t淋巴细胞和b淋巴细胞,以及单核细胞,其每一种都与具有过度促炎免疫应答的炎症性疾病和感染性疾病如呼吸道疾病和肠道疾病相关,在这些疾病的患者中tnfα升高。该实施例还由图10说明。因此,本化合物显示了猪pbmc对细胞因子生成的抑制,表明这样的化合物实施例在炎症性疾病和/或感染性疾病如呼吸道疾病和肠道疾病中的可能的治疗益处。本发明化合物还提供了对动物的瘙痒和疼痛,尤其是动物的急性、慢性、炎症性和神经性疼痛的预防和/或治疗。此外,本发明化合物适用于治疗和/或预防动物的疼痛疾病,尤其是急性、慢性、炎症性和神经性疼痛。所述疾病优选包括痛觉过敏、异常性疼痛、关节炎(例如骨关节炎、类风湿性关节炎和脊柱关节炎)疼痛、经前疼痛、子宫内膜异位-相关的疼痛、手术后疼痛、间质性膀胱炎疼痛、crps(复杂的局部疼痛综合症)、三叉神经痛、前列腺炎疼痛、脊髓损伤引起的疼痛、炎症引起的疼痛、下腰痛、癌痛、化疗-相关的疼痛、hiv-治疗引起的神经病、灼伤引起的疼痛和慢性痛。本发明还提供了一种使用有效量的至少一种本发明的化合物治疗和/或防治动物疾病,尤其是上述疾病的方法。优选提供通过将有效量的至少一种上文定义的本发明式(i)的化合物给药至需要其的动物来治疗和/或预防动物的过敏性和/或炎症性疾病的方法。在本发明上下文中,术语“治疗(treatment)”或“治疗(treating)”包括抑制、延缓、阻止(checking)、缓解、减轻、限制、减少、制止(suppressing)、抵抗或治愈疾病、病状、病症、损伤或健康问题,或所述状态的发展、进程或演进和/或所述状态的症状。术语“疗法(therapy)”在这里理解为与术语“治疗(treatment)”同义。在本发明上下文中,术语“防治(prevention)”、“预防(prophylaxis)”和“阻止(preclusion)”同义使用,并且指的是避免或减少感染、经受、遭受或患有疾病、病状、病症、损伤或健康问题或所述状态的发展或演进和/或所述状态的症状的风险。可以部分或完全治疗或防治疾病、病状、病症、损伤或健康问题。本发明化合物可单独使用,或者如果需要,与其他活性成分结合使用。本发明还提供了含有至少一种本发明化合物和一种或多种其他活性成分的药物,其用于治疗和/或防治动物的过敏性和/或炎症性疾病。适用于组合物的活性成分的优选实例包括:一般可提及活性成分例如抗细菌物质(例如青霉素、万古霉素(vancomycin)、环丙沙星(ciprofloxacin))、抗病毒物质(例如阿昔洛韦(aciclovir)、奥司他韦(oseltamivir))和抗霉菌物质(例如萘替芬(naftifin)、制霉菌素(nystatin))以及γ-球蛋白;免疫调节和免疫抑制化合物如环孢菌素(cyclosporin)、tnf拮抗剂(例如依那西普(etanercept)、英夫利昔(infliximab))、il-1抑制剂(例如阿那白滞素(anakinra)、卡那单抗(canakinumab)、利洛纳塞(rilonacept))、磷酸二酯酶抑制剂(例如阿普斯特(apremilast))、jak/stat抑制剂(例如托法替尼(tofacitinib)、巴瑞替尼(baricitinib)、glpg0634)、来氟米特(leflunomid)、环磷酰胺、利妥昔单抗(rituximab)、贝利木单抗(belimumab)、他克莫司(tacrolimus)、雷帕霉素(rapamycin)、吗替麦考酚酯(mycophenolatemofetil)、干扰素、皮质类固醇(例如强的松(prednisone)、强的松龙(prednisolone)、甲基强的松龙(methylprednisolone)、氢化可的松(hydrocortisone)、倍他米松(betamethasone))、环磷酰胺、咪唑硫嘌呤(azathioprine)和柳氮磺胺吡啶(sulfasalazine);扑热息痛(paracetamol)、非甾体(non-steroidal)抗炎物质(nsaids)(阿司匹林、布洛芬(ibuprofen)、萘普生(naproxen)、依托度酸(etodolac)、塞来昔布(celecoxib)、秋水仙碱(colchicine))。除上述那些之外,本发明irak4抑制剂还可与以下活性成分结合:用于治疗肺部疾病的物质,例如β-2-拟交感神经药(sympathomimetics)(例如舒喘灵(salbutamol))、抗胆碱能药物(anticholinergics)(例如格隆溴铵(glycopyrronium))、甲基黄嘌呤(methylxanthines)(例如茶碱(theophylline))、白三烯(leukotriene)受体拮抗剂(例如孟鲁司特(montelukast))、pde-4(4型磷酸二酯酶(phosphodiesterase))抑制剂(例如罗氟司特(roflumilast))、氨甲蝶呤、ige抗体、咪唑硫嘌呤和环磷酰胺、含皮质醇的制剂;用于治疗骨关节炎(osteoarthritis)的物质,例如非甾体抗炎物质(nsaids)。除了所述两种疗法,对于类风湿性疾病,例如类风湿性关节炎、脊柱关节炎和幼年特发性关节炎,应提及用于b-细胞和t-细胞疗法的氨甲蝶呤和生物制剂(例如利妥昔单抗(rituximab)、阿巴西普(abatacept))。神经营养物质例如乙酰胆碱酯酶抑制剂(例如多奈哌齐(donepezil))、mao(单胺氧化酶(monoaminooxidase))抑制剂(例如司来吉兰(selegiline))、干扰素和镇痉挛剂(anticonvulsives)(例如加巴喷丁(gabapentin));用于治疗心血管疾病的活性成分,例如β阻断剂(例如美托洛尔(metoprolol))、ace抑制剂(例如贝那普利(benazepril))、血管紧张素受体阻断剂(angiotensinreceptorblockers)(例如氯沙坦(losartan)、缬沙坦(valsartan))、利尿剂(diuretics)(例如氢氯噻嗪)、钙通道阻断剂(例如硝苯地平(nifedipine))、他汀类(statins)(例如辛伐他汀(simvastatin)、氟伐他汀(fluvastatin));抗糖尿病药,例如二甲双胍(metformin)、格列奈类(glinides)(例如那格列奈(nateglinide))、dpp-4(二肽基肽酶-4(dipeptidylpeptidase-4))抑制剂(例如利拉利汀(linagliptin)、沙格列汀(saxagliptin)、西格列汀(sitagliptin)、维格列汀(vildagliptin))、sglt2(钠/葡萄糖协同转运蛋白(cotransporter)2)抑制剂/格列净(gliflozin)(例如达格列净(dapagliflozin)、依帕列净(empagliflozin))、肠降血糖素类似物(incretinmimetics)(荷尔蒙葡萄糖依赖性促胰岛素多肽(hormoneglucose-dependentinsulinotropicpeptide)(gip)和类高血糖素样肽1(glucagon-likepeptid1)(glp-1)类似物/激动剂)(例如艾塞那肽(exenatide)、利拉鲁肽(liraglutide)、利西拉肽(lixisenatide))、α-葡萄糖苷酶抑制剂(α-glucosidaseinhibitors)(例如阿卡波糖(acarbose)、米格列醇(miglitol)、伏格列波糖(voglibiose))和磺脲类(例如格列本脲(glibenclamide)、甲苯磺丁脲(tolbutamide))、胰岛素增敏剂(insulinsensitizers)(例如吡格列酮(pioglitazone))和胰岛素疗法(insulintherapy)(例如低精蛋白胰岛素(nphinsulin)、赖脯胰岛素(insulinlispro))。用于治疗慢性炎症性肠道疾病的活性成分,例如美沙拉嗪(mesalazine)、柳氮磺胺吡啶(sulfasalazine)、咪唑硫嘌呤、6-巯基嘌呤或氨甲蝶呤、益生菌(mutaflor、鼠李糖乳杆菌(lactobacillusgg)、植物乳杆菌(lactobacillusplantarum)、嗜酸乳杆菌(l.acidophilus)、干酪乳杆菌(l.casei)、婴儿双歧杆菌(bifidobacteriuminfantis)35624、屎肠球菌(enterococcusfecium)sf68、长双歧杆菌(bifidobacteriumlongum)、大肠杆菌(escherichiacoli)nissle1917);抗生素,例如环丙沙星(ciprofloxacin)和甲硝唑(metronidazole);止泻药,例如洛哌丁胺(loperamide);或通便剂(laxatives)(比沙可啶(bisacodyl))。用于治疗红斑狼疮的免疫抑制剂例如糖皮质激素(glucocorticoids)和非甾体抗炎物质(nsaids)、可的松(cortisone)、氯喹(chloroquine)、环孢菌素(cyclosporine)、咪唑硫嘌呤、贝利木单抗、利妥昔单抗、环磷酰胺。对于皮肤病,维他命d3类似物,例如钙泊三醇(calcipotriol)、他卡西醇(tacalcitol)或骨化三醇(calcitriol)、水杨酸、尿素、环孢素(ciclosporine)、氨甲喋呤、依法利珠单抗(efalizumab)。还应提及用于治疗和/或预防上述疾病的药物,所述药物包含至少一种本发明的化合物和一种或多种用于本发明用途的其他活性成分,尤其是ep4抑制剂(前列腺素e2受体4抑制剂)、p2x3抑制剂(p2x嘌呤受体3)、ptges抑制剂(前列腺素e合酶抑制剂)或akr1c3抑制剂(醛-酮还原酶家族1成员c3抑制剂)。本发明化合物可系统地和/或局部地作用。为此,可以以合适的方式给药,例如通过口服、肠胃外、肺、鼻、舌下、舌、口腔、直肠、皮肤、透皮或结膜途径,经耳或作为植入物或支架。本发明化合物可以以适合这些给药途径的给药形式给药。口服给药的合适的给药形式为:其根据现有技术作用并且快速和/或以缓和方式释放本发明的化合物,并且其含有结晶和/或无定形和/或溶解形式的本发明的化合物,其例如为片剂(未包衣或包衣的片剂,所述包衣片剂例如具有用控制本发明化合物释放的抗胃液包衣或延迟溶解包衣或不溶包衣)、在口腔中迅速崩解的片剂或薄片/扁圆片、薄片/冻干剂、胶囊剂(例如硬或软明胶胶囊剂)、糖衣片剂、咀嚼剂(例如软咀嚼剂)、颗粒剂、丸剂、粉剂、乳剂、悬浮剂、气溶胶或溶液。肠胃外给药可通过避过再吸收步骤(例如通过静脉内、动脉内、心内、脊柱内或腰椎内途径)或包括吸收(例如肌内、皮下、皮内、经皮或腹膜内途径)而完成。适于肠胃外给药的给药形式包括溶液剂、悬浮剂、乳剂、冻干剂或无菌粉剂形式的注射用和输液用制剂。对于其他给药途径,适合的实例为可吸入药物形式(包括粉雾剂(powderinhalers)、喷雾剂)、滴鼻剂、鼻溶液或喷鼻剂;用于经舌、舌下或口腔给药的片剂、薄片/扁平剂或胶囊;栓剂;耳用或眼用制剂、阴道胶囊剂、水性悬浮剂(洗剂、振荡合剂(shakingmixtures))、亲脂性悬浮剂、软膏剂、乳膏剂、倾倒剂(pour-ons)、经皮治疗系统(例如贴剂)、乳剂(milk)、糊剂、泡沫剂(foams)、喷粉剂(sprinklingpowders)、植入物或支架。优选口服给药或肠胃外给药,尤其是口服给药。本发明化合物可转变为所提及的给药形式。这可以本身已知的方式通过与惰性、无毒、药物学上合适的赋形剂混合而完成。这些赋形剂包括载体(例如微晶纤维素、乳糖、甘露醇)、溶剂(例如液体聚乙二醇)、乳化剂和分散剂或润湿剂(例如十二烷基硫酸钠、聚氧化山梨糖醇油酸酯)、粘合剂(例如聚乙烯吡咯烷酮)、合成聚合物和天然聚合物(例如白蛋白)、稳定剂(例如抗氧化剂,例如抗坏血酸)、着色剂(例如无机颜料,例如氧化铁)和矫味剂(flavorcorrectants)和/或矫臭剂(odourcorrectants)。本发明还提供包含至少一种本发明的化合物,通常还包含一种或多种惰性、无毒、药物学上可接受的赋形剂的药物,用于治疗和/或预防动物的过敏性和/或炎症性疾病的方法中。通常,已发现在肠胃外给药的情况下,有利的是给予约0.001至1mg/kg体重、优选约0.01至0.5mg/kg体重的量,以达到有效的结果。在口服给药的情况下,剂量为约0.01至100mg/kg体重、优选约0.01至20mg/kg体重并且最优选0.1至10mg/kg体重。然而,在某些情况下,可能有必要偏离所规定的量,特别地可随体重、给药途径、对活性成分的个体应答、制剂的性质和给药发生的时间或间隔来调整。因此,在某些情况下,用少于上述最小量的量进行可能是足够的,而在其他情况下,必须超过所提及的上限。在给药量较大的情况下,建议将其分为一天内的若干单独剂量。以下工作实施例说明了本发明。本发明不限于所述实施例。除非另有说明,在以下试验和实施例中的百分比为重量百分比;份数为重量份数。液/液溶液的溶剂比、稀释比和浓度数据在每种情况下基于体积计。化合物的制备本发明化合物的制备通过以下合成方案说明。用于合成本发明化合物的起始物料为羧酸(中间体v3),其市售可得或可通过从文献中获知的途径或类似于从文献中获知的途径制备(参见,例如,europeanjournaloforganicchemistry2003,8,1559–1568;chemicalandpharmaceuticalbulletin,1990,38,9,2446–2458;syntheticcommunications2012,42,658–666;tetrahedron,2004,60,51,11869-11874)(参见,例如合成方案1)。一些羧酸v3可由羧酸酯(中间体v2)开始,通过水解(参见,例如,6-(羟甲基)吡啶-2-羧酸乙酯与氢氧化钠水溶液在甲醇中的反应,wo2004113281)来制备,或者,在叔丁酯的情况下,通过与酸如盐酸或三氟乙酸反应(参见,例如,daltontransactions,2014,43,19,7176–7190)来制备。羧酸v3还可以其碱金属盐的形式使用。中间体v2还可任选地由含有氯、溴或碘作为取代基x1的中间体v1来制备,所述制备通过在一氧化碳气氛中,任选地在高压下,在膦配体(例如1,3-双(二苯基膦基)丙烷)、钯化合物(例如乙酸钯(ii))和碱(例如三乙胺)的存在下,在溶剂(例如二甲基亚砜)中加入乙醇或甲醇反应制备(对于制备方法,参见,例如wo2012112743、wo2005082866、chemicalcommunications(cambridge,england),2003,15,1948–1949、wo200661715)。中间体v1市售可得,或可通过从文献中获知的途径制备。示例性的制备方法在以下文献中有详细记载:wo2012061926;europeanjournaloforganicchemistry,2002,2,327–330;synthesis,2004,10,1619–1624;journaloftheamericanchemicalsociety,2013,135,32,12122–12134;bioorganicandmedicinalchemistryletters,2014,24,16,4039–4043;us2007185058;wo2009117421。合成方案1x1为氯、溴或碘。rd为甲基、乙基、苄基或叔丁基。r4、r5各自如在通式(i)中所定义。类似于wo2008/001883,5-氨基-1h-吲唑-6-羧酸甲酯(中间体2)可根据合成方案2,由1h-吲唑-6-羧酸甲酯(中间体0)开始,通过硝化和在碳载钯的存在下用氢气还原中间体1的硝基而制备。对于由中间体2开始来制备中间体3,可使用从文献中已知的各种偶联试剂(aminoacids,peptidesandproteinsinorganicchemistry,第3卷–buildingblocks,catalysisandcouplingchemistry,andrewb.hughes,wiley,第12章-peptide-couplingreagents,407-442;chem.soc.rev.,2009,38,606)。例如,可使用1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐与以下物质结合作为偶联试剂:1-羟基-1h-苯并三唑水合物(hobt,wo2012107475;bioorg.med.chem.lett.,2008,18,2093)、(1h-苯并三唑-1-基氧基)(二甲基氨基)-n,n-二甲基甲亚铵(methaniminium)四氟硼酸盐(tbtu,cas125700-67-6)、(二甲基氨基)-n,n-二甲基(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基氧基)甲亚铵六氟磷酸盐(hatu,cas148893-10-1)、丙烷膦酸酐(以乙酸乙酯溶液或dmf溶液的形式,cas68957-94-8)或二-1h-咪唑-1-基甲酮(cdi),在每种情况下向反应混合物中加入碱如三乙胺或n-乙基-n-异丙基丙-2-胺。优选使用tbtu和于thf中的n-乙基-n-异丙基丙-2-胺。合成方案2取代基r4、r5各自如在通式(i)中所定义。从中间体3开始,可以制备2-取代吲唑衍生物(中间体4)(参见合成方案3)。为此目的,有用的反应包括与任选取代的烷基氯化物、烷基溴化物、烷基碘化物或4-甲基苯磺酸烷基酯的那些反应。所用的烷基卤化物或4-甲基苯磺酸烷基酯市售可得或可类似于从文献中已知的途径制备(对于4-甲基苯磺酸烷基酯的制备,一个实施例是使适量的醇与4-甲基苯磺酰氯在三乙胺或吡啶的存在下反应;参见,例如bioorganicandmedicinalchemistry,2006,14,124277–4294)。任选地,在使用烷基氯化物或烷基溴化物的情况下,还可加入碱金属碘化物如碘化钾或碘化钠。使用的碱可为例如碳酸钾、碳酸铯或氢化钠。在反应性烷基卤化物的情况下,在某些情况下还可使用n-环己基-n-甲基环己胺。有用的溶剂包括,例如1-甲基吡咯烷-2-酮、dmf、dmso或thf。任选地,所使用的烷基卤化物或4-甲基苯磺酸烷基酯可能具有任选地预先已用保护基团保护的官能团(还参见p.g.m.wuts,t.w.greene,greene’sprotectivegroupsinorganicsynthesis,第四版,isbn:9780471697541)。例如,如果使用具有一个或多个羟基基团的烷基卤化物或4-甲基苯磺酸烷基酯,则这些羟基基团可任选地被叔丁基(二甲基)甲硅烷基或本领域技术人员所熟悉的类似的含硅保护基团保护。或者,也可用四氢-2h-吡喃(thp)基团或乙酰基或苯甲酰基来保护羟基基团。然后,可在合成中间体4之后或在合成(i)之后,脱去所使用的保护基团。例如,如果将叔丁基(二甲基甲硅烷基)基团用作保护基团,则可使用于溶剂如thf中的四丁基氟化铵脱去。例如,可使用4-甲基苯磺酸(任选地以一水合物形式)脱去thp保护基团。乙酰基或苯甲酰基可通过用氢氧化钠水溶液处理脱去。任选地,所使用的烷基卤化物或4-甲基苯磺酸烷基酯可含有可通过本领域技术人员已知的氧化或还原反应进行转化的官能团(参见,例如scienceofsynthesis,georgthiemeverlag)。例如,如果官能团为硫化物基团(sulphidegroup),则可通过文献中已知的方法将其氧化为亚砜基或砜基。在亚砜基的情况下,其又可被氧化为砜基。对于这些氧化步骤,可使用例如3-氯过苯甲酸(cas937-14-4)(对此,还参见例如us201094000,其中2-(甲基硫烷基)乙基-1h-吡唑衍生物氧化为2-(甲基亚磺酰基)乙基-1h-吡唑衍生物,以及另外2-(甲基硫烷基)乙基-1h-吡唑衍生物氧化为2-(甲基磺酰基)乙基-1h-吡唑衍生物)。如果所使用的烷基卤化物或对甲苯磺酸酯含有酮基,则可其通过本领域技术人员已知的还原方法还原为醇基(参见,例如chemischeberichte,1980,113,1907–1920中关于硼氢化钠的使用)。这些氧化或还原步骤可在合成中间体4之后或在合成本发明通式(i)的化合物之后进行。或者,可通过中间体3与任选取代的烷基醇的mitsunobu反应(参见,例如k.c.k.swamy等,chem.rev.2009,109,2551-2651)来制备中间体4。可结合使用各种膦化合物如三苯基膦、三丁基膦或1,2-二苯基膦基乙烷与文献中提及的偶氮二羧酸二异丙酯(cas2446-83-5)或其他二氮烯衍生物(k.c.k.swamy等人,chem.rev.2009,109,2551–2651)。优选使用三苯基膦和偶氮二羧酸二异丙酯。如果烷基醇带有官能团,则可以——如上述与烷基卤化物的反应的情况——进行已知的保护基团策略(其他指示可参见p.g.m.wuts,t.w.greene,greene’sprotectivegroupsinorganicsynthesis,第四版,isbn:9780471697541),并且——如上述与烷基卤化物的反应的情况——对中间体4的合成,或在合成本发明通式(i)的化合物之后相应进行氧化和还原步骤。本发明通式(i)的化合物,其中r2和r3定义为c1-c6-烷基(其中r2和r3具有相同的定义),可由中间体4通过格氏反应(grignardreaction)获得(参见,例如ep2489663中1h-吲唑-6-羧酸甲酯衍生物与甲基溴化镁的反应)。对于格氏反应,可使用烷基卤化镁。特别优选于thf或二乙醚中或于thf和二乙醚的混合物中的甲基氯化镁或甲基溴化镁。或者,本发明通式(i)的化合物,其中r2和r3定义为c1-c6-烷基(其中r2和r3具有相同的定义),可由中间体4通过与烷基锂试剂反应(参见,例如wo2006116412中2-氨基-4-氯-1-甲基-1h-苯并咪唑-7-羧酸甲酯衍生物与异丙基锂或叔丁基锂的反应)获得。从中间体4开始,通过用在thf中的氢化铝锂、在thf中的硼氢化锂或在thf中的硼氢化钠(任选添加甲醇)、或硼氢化锂与硼氢化钠的混合物还原,可以制备其中r2和r3定义为h的本发明通式(i)的化合物。合成方案3取代基r1、r2、r3、r4、r5各自如在通式(i)中所定义。中间体5,其中r2和r3定义为c1-c6-烷基(其中r2和r3具有相同的定义),可由中间体3通过格氏反应(参见,例如合成方案4)获得。为此目的,可使用合适的烷基卤化镁,例如于thf或二乙醚中或于thf和二乙醚的混合物中的甲基氯化镁或甲基溴化镁。然后,可由中间体5制备本发明化合物(i)的一部分(i-a),其中r2和r3定义为c1-c6-烷基(其中r2和r3具有相同的定义)。为此目的,类似于合成方案3(中间体3的制备),有用的反应是中间体5与任选取代的烷基氯化物、烷基溴化物、烷基碘化物或4-甲基苯磺酸烷基酯的那些反应。可使用类似于在合成方案3中记载的那些的保护基团策略。或者,为制备其中r2和r3定义为c1-c6-烷基(其中r2和r3具有相同的定义)的本发明化合物(i)的一部分(i-a),可使用中间体5与任选取代的烷基醇的mitsunobu反应(类似于合成方案3)。如果式(i-a)的化合物中的r1包括合适的官能团,则随后可任选地类似于合成方案3,使用氧化或还原反应制备其他本发明化合物。合成方案4取代基r1、r4、r5分别如在通式(i)中所定义。r2和r3总是具有相同的定义,并且都为c1-c6-烷基。可以以可替代的方式由中间体1制备中间体4(参见合成方案5)。首先,通过如合成方案3(由中间体3制备中间体4)中的方法将中间体1转化为中间体6。然后,中间体6可通过硝基的还原而转化为中间体7。例如,硝基可用碳载钯在氢气气氛下还原(参见,例如wo2013174744中关于将6-异丙氧基-5-硝基-1h-吲唑还原为6-异丙氧基-1h-吲唑-5-胺),或用在水和乙醇中的铁和氯化铵还原(请参见,例如journalofthechemicalsociety,1955,2412-2419),或使用氯化锡(ii)(cas7772-99-8)还原。优选使用在水和乙醇中的铁和氯化铵。由中间体7制备中间体4可类似于合成方案2进行(由中间体2制备中间体3)。如合成方案3所述,在合成方案5的情况下,也可任选地使用保护基团策略。从中间体6或中间体7开始,任选地也可以如合成方案3所描述的,进行本领域技术人员已知的氧化或还原反应(参见,例如scienceofsynthesis,georgthiemeverlag)。合成方案5取代基r1、r4、r5各自如在通式(i)中所定义。实施例化合物的合成缩写和说明dmfn,n-二甲基甲酰胺dmso二甲基亚砜thf四氢呋喃rt室温hplc高效液相色谱法h小时hcooh甲酸mecn乙腈min分钟uplc超高效液相色谱dad二极管阵列检测器elsd蒸发光散射检测器esi电喷雾电离sqd单四极检测器(singlequadrupoledetector)cpg芯移出精密玻璃(core-pulledprecisionglass)nh3氨术语氯化钠溶液总是指饱和氯化钠水溶液。中间体和实施例的化学名称使用acd/labs(batch12.01版)软件生成。方法在某些情况下,本发明化合物及其前体和/或中间体通过lc-ms进行分析。方法a1:uplc(mecn-hcooh):仪器:watersacquityuplc-mssqd3001;柱:acquityuplcbehc181.750×2.1mm;洗脱液a:水+0.1体积%的甲酸(99%),洗脱液b:乙腈;梯度:0-1.6min1-99%b,1.6-2.0min99%b;流率0.8ml/min;温度:60℃;进样量:2μl;dad扫描:210-400nm;msesi+,esi-,扫描范围160-1000m/z;elsd。方法a2:uplc(mecn-nh3):仪器:watersacquityuplc-mssqd3001;柱:acquityuplcbehc181.750×2.1mm;洗脱液a:水+0.2体积%的氨(32%),洗脱液b:乙腈;梯度:0-1.6min1-99%b,1.6-2.0min99%b;流率0.8ml/min;温度:60℃;进样量:2μl;dad扫描:210-400nm;msesi+,esi-,扫描范围160-1000m/z;elsd。方法a3:(lc-ms)仪器:agilent1290infinitylc;柱:acquityuplcbehc181.750×2.1mm;洗脱液a:水+0.05体积%的甲酸,洗脱液b:乙腈+0.05%体积的甲酸;梯度:0-1.7min2-90%b,1.7-2.0min90%b;流率1.2ml/min;温度:60℃;进样量:2μl;dad扫描:190-390nm;ms:agilenttof6230。方法a4:(lc-ms)仪器:watersacquity;柱:kinetex(phenomenex),50×2mm;洗脱液a:水+0.05体积%的甲酸,洗脱液b:乙腈+0.05%体积的甲酸;梯度:0-1.9min1-99%b,1.9-2.1min99%b;流率1.5ml/min;温度:60℃;进样量:0.5μl;dad扫描:200-400nm。在某些情况下,本发明化合物和其前体和/或中间体通过以下说明性的制备型hplc方法纯化:方法p1:系统:waters自动纯化系统:泵2545,样品管理器2767,cfo,dad2996,elsd2424,sqd;柱:xbridgec185μm100×30mm;洗脱液a:水+0.1体积%的甲酸,洗脱液b:乙腈;梯度:0-8min10-100%b,8-10min100%b;流率:50ml/min;温度:室温;溶液:最大250mg/最大2.5mldmso或dmf;进样量:1×2.5ml;检测:dad扫描范围210–400nm;msesi+,esi-,扫描范围160-1000m/z。方法p2:系统:waters自动纯化系统:泵254,样品管理器2767,cfo,dad2996,elsd2424,sqd3100;柱:xbridgec185μm10×30mm;洗脱液a:水+0.2体积%的氨(32%),洗脱液b:甲醇;梯度:0-8min30-70%b;流率:50ml/min;温度:室温;检测:dad扫描范围210–400nm;msesi+,esi-,扫描范围160-1000m/z;elsd。方法p3:系统:labomatic,泵:hd-5000,馏分收集器:labocolvario-4000,uv检测器:knaueruvd2.1s;柱:xbridgec185μm100×30mm;洗脱液a:水+0.2体积%的氨(25%),洗脱液b:乙腈;梯度:0-1min15%b,1-6.3min15-55%b,6.3-6.4min55-100%b,6.4-7.4min100%b;流率:60ml/min;温度:室温;溶液:最大250mg/2mldmso;进样量:2×2ml;检测:uv218nm;软件:scpaprepcon5。方法p4:系统:labomatic,泵:hd-5000,馏分收集器:labocolvario-4000,uv检测器:knaueruvd2.1s;柱:chromatorexrpc1810μm125×30mm;洗脱液a:水+0.1%体积的甲酸,洗脱液b:乙腈;梯度:0-15min65–100%b;流率:60ml/min;温度:室温;溶液:最大250mg/2mldmso;注射:2×2ml;检测:uv254nm;软件:scpaprepcon5。方法p5:系统:sepiatec:prepsfc100,柱:chiralpakia5μm250×20mm;洗脱液a:二氧化碳,洗脱液b:乙醇;梯度:等度20%b;流率:80ml/min;温度:40℃;溶液:最大250mg/2mldmso;进样量:5×0.4ml;检测:uv254nm。方法p6:系统:agilent:prep1200,2xprep泵,dla,mwd,gilson:liquidhandler215;柱:chiralceloj-h5μm250×20mm;洗脱液a:己烷,洗脱液b:乙醇;梯度:等度30%b;流率:25ml/min;温度:25℃;溶液:187mg/8ml乙醇/甲醇;进样量:8×1.0ml;检测:uv280nm。方法p7:系统:labomatic,泵:hd-5000,馏分收集器:labocolvario-4000,uv检测器:knaueruvd2.1s;柱:xbridgec185μm100×30mm;洗脱液a:水+0.1%体积的甲酸,洗脱液b:乙腈;梯度:0-3min:65%b等度,3-13min:65-100%b;流率:60ml/min;温度:室温;溶液:最大250mg/2mldmso;进样量:2×2ml;检测:uv254nm。方法p8:系统:agilent:prep1200,2×prep泵,dla,mwd,gilson:liquidhandler215;柱:chiralpakif5μm250×20mm;洗脱液a:乙醇,洗脱液b:甲醇;梯度:等度50%b;流率:25ml/min;温度:25℃;溶液:600mg/7mln,n-二甲基甲酰胺:进样量:10×0.7ml;检测:uv254nm。在某些情况下,物质混合物通过硅胶柱层析法纯化。为制备某些本发明化合物及其前体和/或中间体,使用来自biotage的设备在硅胶上进行柱色谱纯化(“快速色谱法”)。这使用购自biotage的小柱完成,例如不同尺寸的“snapcartridge,kp_sil”小柱和购自interchim的不同尺寸的“interchimpuriflashsilicahp15umflashcolumn”小柱。起始物料中间体v2-16-(2-羟基丙-2-基)吡啶-2-羧酸甲酯将2.00g(9.26mmol)的2-(6-溴吡啶-2-基)丙-2-醇(cas638218-78-7)溶于20ml的甲醇和20ml的dmso中。随后,加入250mg的1,3-双(二苯基膦基)丙烷、130mg的乙酸钯(ii)和3ml的三乙胺。在室温下将反应混合物用一氧化碳吹扫三次,并在13巴的一氧化碳气氛中搅拌30min。通过施加真空除去一氧化碳气氛,并在100℃下将混合物在14巴的一氧化碳气氛中搅拌24h。使高压釜减压,向反应混合物中加入水,并用乙酸乙酯萃取反应混合物三次,用饱和碳酸氢钠水溶液和氯化钠水溶液洗涤,通过疏水性过滤器过滤并浓缩。获得1.60g的粗产物。uplc-ms(方法a1):rt=0.76min(uv检测器:tic),分子量测定值195.00。中间体v3-16-(2-羟基丙-2-基)吡啶-2-羧酸钾首先将1.60g的中间体0-1的粗产物加入15ml的甲醇中,加入0.74g的氢氧化钾,并将混合物在50℃下搅拌16.5h。浓缩后,获得2.1g的残余物,其无需进一步纯化即可使用。uplc-ms(方法a1):rt=0.47min(uv检测器:tic),分子量测定值181.00。中间体1-15-硝基-1h-吲唑-6-羧酸甲酯在具有cpg搅拌器、滴液漏斗和内部温度计的三口烧瓶中,将4.60g(26.1mmol)的1h-吲唑-6-羧酸甲酯(cas号:170487-40-8)溶于120ml的硫酸(96%)中,并冷却至-15℃。在15min内,向该溶液中逐滴加入预先制备并冷却的硝化酸(10ml的96%硫酸于5ml65%的硝酸中)。滴加完成后,将混合物再搅拌1h(内部温度为-13℃)。将反应混合物加入到冰中,抽滤出沉淀物,用水洗涤并在干燥箱中在50℃下减压干燥。获得5.49g的标题化合物。uplc-ms(方法a2):rt=0.75minms(esipos):m/z=222(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=3.87(s,3h),7.96(s,1h),8.44(s,1h),8.70(s,1h),13.98(br.s.,1h)。中间体2-15-氨基-1h-吲唑-6-甲酸甲酯将4.40g(19.8mmol)的5-硝基-1h-吲唑-6-羧酸甲酯(中间体1-1)溶于236ml的甲醇中,并在标准氢气压力下用1.06g(0.99mmol)活性炭载钯在25℃下氢化3h。将反应混合物通过硅藻土过滤,用甲醇洗涤过滤物,并浓缩滤液。获得3.53g的标题化合物。1hnmr(300mhz,dmso-d6):δ[ppm]=3.85(s,3h)6.01(s,2h)6.98(s,1h)7.79-7.91(m,1h)7.99(s,1h)12.84(br.s.,1h)。中间体3-15-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯首先将4.95g(25.9mmol)的6-(三氟甲基)吡啶-2-羧酸加入到45mlthf中。加入9.07g(28.2mmol)的四氟硼酸o-(苯并三唑-1-基)-n,n,n′,n′-四甲基脲和4.92ml(28.2mmol)的n-乙基-n-异丙基丙-2-胺,并将混合物在25℃下搅拌30min。随后,加入4.50g(23.5mmol)的5-氨基-1h-吲唑-6-羧酸甲酯(中间体2-1),并将混合物在25℃下搅拌24h。反应混合物通过膜过滤器抽滤,固体用thf和水洗涤,并在干燥箱中干燥过夜。获得7.60g标题化合物。uplc-ms(方法a2):rt=1.16minms(esipos):m/z=365(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=3.97(s,3h),8.13-8.27(m,2h),8.30(s,1h),8.33-8.45(m,1h),8.45-8.51(m,1h),9.15(s,1h),12.57(s,1h),13.44(s,1h)。中间体3-25-({[6-(二氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯首先将2.85g(23.5mmol)的6-(二氟甲基)吡啶-2-羧酸加入到30ml的thf中。加入6.05g(18.8mmol)四氟硼酸o-(苯并三唑-1-基)-n,n,n′,n′-四甲基脲和3.3ml的n-乙基-n-异丙基丙-2-胺,并将混合物在室温下搅拌10min。随后,加入3.00g(15.7mmol)的5-氨基-1h-吲唑-6-羧酸甲酯,并将混合物在室温下搅拌过夜。将反应混合物与水混合,抽滤出沉淀物,并用水和二氯甲烷反复洗涤。获得1.53g(理论值的27%)标题化合物。分离滤液的各相,将有机相浓缩,与少量的二氯甲烷混合并悬浮于超声浴中,并抽滤出沉淀物。这再得到1.03g标题化合物。1h-nmr(第一产物部分,300mhz,dmso-d6):δ[ppm]=3.99(s,3h),7.09(t,1h),8.00(d,1h),8.21-8.40(m,4h),9.14(s,1h),12.53(s,1h),13.44(s,1h)。中间体3-35-({[6-(2-羟基丙-2-基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯首先将2.10g的6-(2-羟基丙-2-基)吡啶-2-羧酸钾(中间体v3-1)加入到15ml的thf中。加入3.69g(11.5mmol)的四氟硼酸o-(苯并三唑-1-基)-n,n,n′,n′-四甲基脲和2.00ml的n-乙基-n-异丙基丙-2-胺,并将混合物在室温下搅拌15min。随后,加入1.83g(9.58mmol)的5-氨基-1h-吲唑-6-羧酸甲酯(中间体2-1),并将混合物在室温下搅拌19h。将混合物与水和乙酸乙酯混合,过滤出未溶解的固体,分离滤液的各相,水相用乙酸乙酯萃取两次,用氯化钠溶液洗涤,通过疏水性过滤器过滤,浓缩,并通过硅胶柱层析法(己烷/乙酸乙酯)纯化。除去溶剂后,获得1.56g黄色泡沫状的标题化合物。uplc-ms(方法a1):rt=1.00min(uv检测器:ticsmooth),分子量测定值354.00。1h-nmr(500mhz,dmso-d6):δ[ppm]=1.63(s,6h),3.97(s,3h),5.37(s,1h),7.90-7.95(m,1h),8.03-8.07(m,2h),8.23(s,1h),8.29(s,1h),9.19(s,1h),12.79(s,1h),13.41(br.s.,1h)。中间体4-12-(氧杂环丁烷-3-基甲基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯将1.00g(2.66mmol)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)溶于10ml的dmf中,并且在加入1.10g(7.99mmol)的碳酸钾和221mg(1.33mmol)的碘化钾之后,将混合物在25℃下搅拌30min。加入603mg(3.99mmol)的3-溴甲基氧杂环丁烷,并将混合物在25℃下搅拌24h。将反应混合物在水和乙酸乙酯之间分层。混合物用乙酸乙酯萃取两次,并将合并的有机相通过疏水性过滤器过滤,浓缩。将残余物通过硅胶柱色谱纯化法(己烷/乙酸乙酯)纯化。获得260mg的标题化合物。uplc-ms(方法a2):rt=1.24minms(esipos):m/z=435(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=3.49-3.64(m,1h),3.95(s,3h),4.49(t,2h),4.68(dd,2h),4.81(d,2h),8.20(dd,1h),8.35-8.41(m,1h),8.43-8.49(m,2h),8.55-8.58(m,1h),9.06(s,1h),12.53(s,1h)。中间体4-22-(2-甲氧基乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯将1.00g(2.75mmol)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)溶于5ml的dmf中,并在搅拌的同时加入387μl(4.12mmol)的2-溴乙基甲基醚、1.14g(8.23mmol)的碳酸钾和228mg(1.37mmol)的碘化钾。将反应混合物在25℃下搅拌24h,用水稀释,并用乙酸乙酯萃取两次。将合并的有机相通过疏水性过滤器过滤,浓缩。残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得12mg的标题化合物。uplc-ms(方法a1):rt=1.24minms(esipos):m/z=423(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=3.24(s,3h),3.86(t,2h),3.96(s,3h),4.65(t,2h),8.21(dd,1h),8.35-8.42(m,1h),8.43-8.51(m,2h),8.52(d,1h),9.06(s,1h),12.53(s,1h)。中间体4-32-(3-甲氧基丙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯将1.00g(2.75mmol)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)溶于5ml的dmf中,并在搅拌的同时加入460μl(4.12mmol)的1-溴-3-甲氧基丙烷、1.14g(8.23mmol)的碳酸钾和228mg(1.37mmol)的碘化钾。将反应混合物在25℃下搅拌72h,用水稀释,并用乙酸乙酯萃取两次。将合并的有机相通过疏水性过滤器过滤,浓缩。残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得28mg的标题化合物。uplc-ms(方法a1):rt=1.29minms(esipos):m/z=437(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=2.17(五重峰,2h),3.24(s,3h),3.33-3.36(m,2h),3.96(s,3h),4.53(t,2h),8.21(dd,1h),8.35-8.42(m,1h),8.45-8.49(m,2h),8.54(d,1h),9.06(s,1h),12.54(s,1h)。中间体4-42-(3-羟基-3-甲基丁基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯制备方法1首先将930mg(2.55mmol)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)、1.06g的碳酸钾和212mg的碘化钾溶于9ml的dmf中,并将混合物搅拌15min。然后,加入0.62ml的4-溴-2-甲基丁-2-醇,并将混合物在60℃下搅拌16h。将混合物与水混合,并用乙酸乙酯萃取两次,将萃取物用饱和氯化钠溶液洗涤三次,过滤并浓缩。硅胶柱层析法(己烷/乙酸乙酯)纯化获得424mg的标题化合物。uplc-ms(方法a2):rt=1.21min(uv检测器:tic),分子量测定值450.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.16(s,6h)2.02-2.11(m,2h)3.96(s,3h)4.51-4.60(m,3h)8.20(dd,j=7.83,1.01hz,1h)8.39(s,1h)8.45(s,2h)8.55(d,j=0.76hz,1h)9.05(s,1h)12.52(s,1h)。制备方法2首先将1.95g(7.03mmol)的5-氨基-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体7-1)加入到30ml的thf中。加入1.48g(7.37mmol)的6-(三氟甲基)吡啶-2-羧酸、2.71g(8.44mmol)的四氟硼酸o-(苯并三唑-1-基)-n,n,n′,n′-四甲基脲和1.47ml(8.44mmol)的n-乙基-n-异丙基丙-2-胺,并将混合物在25℃下搅拌20.5h。加入水,混合物用乙酸乙酯萃取三次,萃取物用氯化钠溶液洗涤,通过疏水性过滤器过滤并浓缩。残余物通过硅胶柱层析法(己烷/乙酸乙酯梯度)分离。获得2.79g的标题化合物。uplc-ms(方法a1):rt=1.23min(uv检测器:tic),分子量测定值450.00。中间体4-52-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯首先将1.00g(2.66mmol,97%)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)加入到50ml的dmf中,在搅拌的同时加入1.10g(7.99mmol)的碳酸钾和221mg(1.33mmol)的碘化钾,将混合物在25℃下搅拌30min。随后,加入857μl(3.99mmol)的(2-溴乙氧基)(叔丁基)二甲基硅烷,并将混合物在25℃下搅拌24h。将反应混合物用水稀释,并用乙酸乙酯萃取。将合并的有机相通过疏水性过滤器过滤并浓缩。残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得400mg的标题化合物。uplc-ms(方法a1):rt=1.58minms(esipos):m/z=523(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=-0.18--0.13(m,6h),0.74(s,9h),3.96(s,3h),4.08(t,2h),4.57(t,2h),8.15-8.25(m,1h),8.32-8.43(m,1h),8.43-8.52(m,3h),9.07(s,1h),12.53(s,1h)。中间体4-62-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯与中间体4-5类似地,将1.00g(2.75mmol)的5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)溶于10ml的dmf中,在搅拌的同时加入1.14g(8.24mmol)的碳酸钾和228mg(1.37mmol)的碘化钾,并将混合物在25℃下搅拌30min。随后,加入1.04g(4.12mmol)的(3-溴丙氧基)(叔丁基)二甲基硅烷,并将混合物在25℃下搅拌24h。过滤反应混合物,并用乙酸乙酯洗涤滤饼。使反应混合物在水和乙酸乙酯之间分层,水相用乙酸乙酯萃取两次。将合并的有机相通过疏水性过滤器过滤,并浓缩。通过制备型hplc纯化残余物,获得428mg的标题化合物。uplc-ms(方法a1):rt=1.63minms(esipos):m/z=537(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=-0.02-0.06(m,6h),0.87(s,9h),2.14(quin,2h),3.62(t,2h),3.96(s,3h),4.54(t,2h),8.20(d,1h),8.35-8.42(m,1h),8.43-8.48(m,3h),8.49-8.53(m,1h),9.06(s,1h)。中间体4-75-({[6-(2-羟基丙-2-基)吡啶-2-基]羰基}氨基)-2-(4,4,4-三氟丁基)-2h-吲唑-6-羧酸甲酯首先将300mg(0.80mmol)的5-({[6-(2-羟基丙-2-基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-3)加入到4.5ml的dmf中。加入287mg(1.21mmol)的1,1,1-三氟-4-碘代丁烷和333mg的碳酸钾,并将混合物在100℃下搅拌23h。加入水,混合物用乙酸乙酯萃取三次。浓缩该混合物,并通过制备型hplc纯化产物。获得72mg的标题化合物。uplc-ms(方法a1):rt=1.26min(uv检测器:tic),分子量测定值464.17。中间体4-85-{[(5-氟-6-甲基吡啶-2-基)羰基]氨基}-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯类似于中间体4-4(制备方法2),使195mg(0.46mmol)的5-氨基-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体7-1)与78mg(0.50mmol)的5-氟-6-甲基吡啶-2-羧酸反应19.5h。在类似的水处理后,获得228mg的粗产物。uplc-ms(方法a1):rt=1.20min(uv检测器:tic),分子量测定值414.00。中间体4-92-(3-羟基-3-甲基丁基)-5-{[(6-甲基吡啶-2-基)羰基]氨基}-2h-吲唑-6-羧酸甲酯类似于中间体4-4(制备方法2),使195mg(0.46mmol)的5-氨基-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体7-1)与70mg(0.50mmol)的6-甲基吡啶-2-羧酸反应19.5h。在类似的水处理后,获得278mg的标题化合物作为粗产物。uplc-ms(方法a1):rt=1.14min(uv检测器:tic),质量测定值396.00。中间体4-102-[3-(2,2,2-三氟乙氧基)丙基]-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯将于3mldmf中的250mg(0.58mmol)5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-1)、193mg(0.88mmol)3-溴丙基-2,2,2-三氟乙基醚、242mg碳酸钾和145mg碘化钾的混合物在100℃下搅拌20h。加入水,用乙酸乙酯萃取混合物,并将萃取物用氯化钠溶液洗涤,并浓缩。通过制备型hplc纯化,获得52mg的标题化合物。uplc-ms(方法a1):rt=1.39min(uv检测器:tic),分子量测定值504.12。中间体4-115-({[6-(二氟甲基)吡啶-2-基]羰基}氨基)-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯首先将2.00g的5-氨基-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体7-1)加入到40ml的thf中。加入1.50g的6-(二氟甲基)吡啶-2-羧酸、2.78g的四氟硼酸o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲(tbtu,cas号125700-67-6)和1.5ml的n-乙基-n-异丙基丙-2-胺,并将混合物在室温下搅拌24h。加入水,将混合物用乙酸乙酯萃取三次,合并的有机相用氯化钠溶液洗涤并通过疏水性过滤器过滤。浓缩混合物,并将残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得3.05g黄色固体状的标题化合物。uplc-ms(方法a1):rt=1.15min(uv检测器tic),分子量测定值432.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.17(s,6h),2.04-2.11(m,2h),3.99(s,3h),4.52-4.60(m,3h),7.10(t,1h),8.00(dd,1h),8.28-8.38(m,2h),8.44–8.47(m,1h),8.56(d,1h),9.05(s,1h),12.49(s,1h)。中间体5-1n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺向在冰水冷却浴中冷却的1.50g(4.12mmol)5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯于20mlthf中的溶液中小心地加入6.9ml(5当量)3m的甲基溴化镁于二乙醚中的溶液。在用冰浴冷却的同时,将混合物搅拌1h,并在室温下搅拌19.5h。另加入2当量的甲基溴化镁溶液,并将混合物在室温下再搅拌24h。加入饱和氯化铵水溶液,搅拌混合物,并用乙酸乙酯萃取三次。将合并的有机相用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得763mg的标题化合物。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.63(s,6h),5.99(s,1h),7.49(s,1h),8.06(s,1h),8.14-8.19(m,1h),8.37(t,1h),8.46(d,1h),8.78(s,1h),12.32(s,1h),12.97(s,1h)。中间体5-26-(二氟甲基)-n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]吡啶-2-羧酰胺类似于中间体5-1的制备,使在10mlthf中的2.40g(6.93mmol)的5-({[6-(二氟甲基)吡啶-2-基]羰基}氨基)-1h-吲唑-6-羧酸甲酯(中间体3-2)与三份3m的甲基溴化镁的二乙醚溶液反应(6.9ml,然后在室温下搅拌45min;11.6ml,然后在室温下搅拌2h;6.9ml,然后在室温下搅拌2h)。进行如对中间体5-1的后处理后,获得2.39g的粗产物,其不需进一步纯化即可使用。中间体6-12-(3-羟基-3-甲基丁基)-5-硝基-2h-吲唑-6-羧酸甲酯首先将5.00g(22.6mmol)的5-硝基-1h-吲唑-6-羧酸甲酯(中间体1-1)加入到40ml的dmf中。加入5.65g(33.9mmol)的4-溴-2-甲基丁-2-醇、9.37g(67.8mmol)的碳酸钾和5.63g(33.9mmol)的碘化钾,并将混合物在100℃下搅拌20h。加入水,混合物用乙酸乙酯萃取三次,将萃取物用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。将残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。将获得的固体与二乙醚一起搅拌,抽滤出,用二乙醚洗涤并干燥。获得2.49g的标题化合物。uplc-ms(方法a1):rt=0.93min(uv检测器:tic),分子量测定值307.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.15(s,6h),2.02-2.11(m,2h),3.84(s,3h),4.54(s,1h),4.58-4.65(m,2h),8.05(s,1h),8.69(s,1h),8.86(s,1h)。中间体7-15-氨基-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯将4.53g的铁和217mg的氯化铵加入到2.49g(8.10mmol)2-(3-羟基-3-甲基丁基)-5-硝基-2h-吲唑-6-羧酸甲酯(中间体6-1)于30ml乙醇和10ml水的溶液中,并将混合物在90℃下搅拌21.5h。混合物通过硅藻土过滤,并用乙醇洗涤三次,浓缩滤液,并将残余物与水混合。萃取用乙酸乙酯进行三次(为改善相分离,加入氯化钠溶液)。将合并的有机相用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。获得1.95g(理论值的85%)的标题化合物。uplc-ms(方法a1):rt=0.67min(uv检测器:tic),分子量测定值277.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.14(s,6h),1.96-2.08(m,2h),3.85(s,3h),4.39-4.51(m,3h),5.81(s,2h),6.80(s,1h),8.05(s,1h),8.18(s,1h)。工作实施例实施例1n-[6-(2-羟基丙-2-基)-2-(2-甲氧基乙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将75mg(0.18mmol)的2-(2-甲氧基乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-2)溶于500μl的thf中,并与887μl(0.89mmol)的1m的甲基溴化镁的thf溶液混合。将反应混合物在25℃下搅拌60min。随后,小心地加入1ml的饱和氯化铵水溶液,并过滤混合物。水相用乙酸乙酯萃取两次,合并有机相,通过疏水过滤器过滤,并浓缩。将残余物溶于3ml的dmso中,并通过制备型hplc纯化。将含有产物的部分冷冻干燥。获得20mg的标题化合物。uplc-ms(方法a1):rt=1.08minms(esipos):m/z=423(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=1.62(s,6h),3.22(s,3h),3.82(t,2h),4.55(t,2h),5.96(s,1h),7.57(s,1h),8.16(d1h),8.29-8.42(m,2h),8.42-8.50(m,1h),8.71(s,1h),12.36(s,1h)。实施例2n-[6-(羟甲基)-2-(2-甲氧基乙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将13mg(0.36mmol)的氢化铝锂悬浮于1ml的thf中,并将混合物冷却至0℃。逐滴加入75mg(0.17mmol)的2-(2-甲氧基乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-2)溶于500μlthf中的溶液,并将混合物在25℃下搅拌60min。用水稀释该混合物并用乙酸乙酯萃取两次,合并的有机相用氯化钠溶液洗涤,通过疏水性过滤器过滤,浓缩,并在减压下干燥。获得13mg的标题化合物。uplc-ms(方法a2):rt=0.99minms(esipos):m/z=394(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=3.23(s,3h),3.83(t,2h),4.56(t,2h),4.69(d,2h),5.77(t,1h),7.57(s,1h),8.19(d,1h),8.33-8.41(m,2h),8.43-8.47(m,1h),8.51(s,1h),11.20(s,1h)。实施例3n-[6-(2-羟基丙-2-基)-2-(3-甲氧基丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将75mg(0.17mmol)的2-(3-甲氧基丙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-3)溶于500μl的thf中,并与859μl(0.86mmol)的1m的甲基溴化镁的thf溶液混合。将反应混合物在25℃下搅拌60min。随后,小心地加入1ml饱和氯化铵溶液,并过滤混合物。水相用乙酸乙酯萃取两次,合并有机相,通过疏水过滤器过滤,并浓缩。将残余物溶于3ml的dmso中,并通过制备型hplc纯化。将含有产物的部分冷冻干燥。得到25mg的标题化合物。uplc-ms(方法a1):rt=1.13minms(esipos):m/z=437(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=1.62(s,6h),2.14(quin,2h),3.23(s,3h),3.26-3.32(m,2h),4.44(t,2h),5.95(s,1h),7.58(s,1h),8.16(d,1h),8.31-8.40(m,2h),8.43-8.48(m,1h),8.72(s,1h),12.36(s,1h)。实施例4n-[6-(羟甲基)-2-(3-甲氧基丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将13mg的氢化铝锂悬浮于thf中,并将混合物冷却至0℃。逐滴加入75mg(0.17mmol)的2-(3-甲氧基丙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-3)于thf中的溶液,并使混合物在30min内达到室温。用水稀释该混合物并过滤,残余物用乙酸乙酯洗涤,并用乙酸乙酯萃取滤液。将合并的乙酸乙酯相用氯化钠溶液洗涤,通过疏水过滤器过滤,浓缩。将残余物通过制备型hplc纯化。1hnmr(300mhz,dmso-d6):δ[ppm]=2.14(quin,2h),3.23(s,3h),3.29(t,2h),4.45(t,2h),4.68(d,2h),5.77(t,1h),7.58(s,1h),8.18(d,1h),8.32-8.48(m,3h),8.51(s,1h),11.21(s,1h)。实施例5n-[2-(2-羟乙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺步骤a:n-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺的制备将100mg(0.19mmol)的2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-5)溶于1ml的thf中,并与669μl(0.67mmol)的1m的甲基溴化镁的thf溶液混合。将反应混合物在25℃下搅拌60min。另加入287μl(0.29mmol)的1m的甲基溴化镁的thf溶液,将混合物在25℃下搅拌3h。随后,小心地加入20ml的饱和氯化铵溶液,并过滤混合物。水相用乙酸乙酯萃取两次,合并有机相,用硫酸镁干燥,过滤,浓缩,并在减压下干燥。获得50mgn-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺。uplc-ms(方法a2):rt=1.51minms(esipos):m/z=523(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=-0.17--0.09(m,6h),0.78(s,9h),1.62(s,6h),4.04(t,2h),4.47(t,2h),5.98(s,1h),7.57(s,1h),8.16(d,1h),8.29(s,1h),8.37(t,1h),8.45(d,1h),8.73(s,1h),12.38(s,1h)。步骤b:将50mg(96μmol)的n-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺溶于1.0ml的thf中,并与144μl(0.14mmol)的1m的四丁基氟化铵的thf溶液混合。将反应混合物在室温下搅拌1h。用水稀释该混合物并用乙酸乙酯萃取两次,将合并的有机相用饱和氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。获得36mg的n-[2-(2-羟乙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(实施例5)。1h-nmr(400mhz,dmso-d6):d[ppm]=1.62(s,6h),3.86(q,2h),4.43(t,2h),4.95(t,1h),5.94(s,1h),7.57(s,1h),8.16(dd,1h),8.30(s,1h),8.37(t,1h),8.45(d,1h),8.72(s,1h),12.36(s,1h)。uplc-ms(方法a2):rt=0.97min(uv检测器:tic),分子量测定值408.00。实施例6n-[6-(2-羟基丙-2-基)-2-(3-羟丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺步骤a:n-[2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺的制备将50mg(0.09mmol)的2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-6)溶于500μl的thf中,并与326μl(0.33mmol)的1m的甲基溴化镁的thf溶液混合。将反应混合物在25℃下搅拌60min。随后,小心地加入20ml的饱和氯化铵溶液,并将混合物用乙酸乙酯萃取两次。将合并的有机相通过疏水过滤器过滤,浓缩,并在减压下干燥。残余物通过制备型hplc纯化。获得40mg的n-[2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺。uplc-ms(方法a1):rt=1.58minms(esipos):m/z=537(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=0.02-0.05(m,6h),0.84-0.91(m,9h),1.62(s,6h),2.02-2.18(m,2h),3.55-3.62(m,2h),4.45(t,2h),5.96(s,1h),7.57(s,1h),8.16(d,1h),8.31(s,1h),8.33-8.42(m,1h),8.45(d,1h),8.72(s,1h),12.37(s,1h)。步骤b:将37mg(0.07mmol)的n-[2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺溶于500μl的thf中,并与207μl(0.21mmol)的1m的四丁基氟化铵的thf溶液混合。将反应混合物在25℃下搅拌2h。用水稀释该混合物并用乙酸乙酯萃取两次,合并的有机相用饱和氯化钠溶液洗涤,过滤并浓缩。通过制备型hplc纯化后,获得10mg的n-[6-(2-羟基丙-2-基)-2-(3-羟丙基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(实施例6,含有次要组分)。uplc-ms(方法a2):rt=1.00minms(esipos):m/z=423(m+h)+1hnmr选定的信号(400mhz,dmso-d6):δ[ppm]=1.61(s),2.00-2.12(m),3.38(t,2h),4.44(t,2h),4.62(br.s.,1h),5.93(br.s.,1h),7.55(s,1h),8.13(d,1h),8.27-8.38(m,2h),8.43(d,1h),8.71(s,1h),12.30(br.s.,1h)。实施例7n-[2-(2-羟乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺步骤a:n-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将100mg(0.19mmol)的2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-5)溶于1ml的thf中,并与191μl(0.38mmol)的2m的硼氢化锂溶液混合。将混合物在25℃下搅拌24h。加入14mg(0.38mmol)的硼氢化钠和500μl甲醇,并将混合物在25℃下搅拌4h。再加入14mg(0.38mmol)的硼氢化钠,并将混合物在25℃下搅拌24h。向反应混合物中小心地加入水,并除去有机相。然后将混合物用乙酸乙酯萃取两次,并将合并的有机相用饱和氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。将残余物溶于2ml的dmso中,并通过制备型hplc纯化。获得30mg的n-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺。uplc-ms(方法a2):rt=1.44minms(esipos):m/z=495(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=-0.16--0.12(m,6h),0.75-0.79(m,9h),4.05(t,2h),4.48(t,2h),4.69(d,2h),5.75-5.77(m,1h),7.57(s,1h),8.18(dd,1h),8.30-8.33(m,1h),8.38(t,1h),8.45(d,1h),8.51(s,1h),11.20(s,1h)。步骤b:将33mg(0.07mmol)的n-[2-(2-{[叔丁基(二甲基)甲硅烷基]氧基}乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺溶于1ml的thf中,并与100μl(0.10mmol)的1m的四丁基氟化铵的thf溶液混合。将反应混合物在25℃下搅拌1h。用水将该混合物稀释,并用乙酸乙酯萃取两次,合并的有机相用饱和氯化钠溶液洗涤,通过疏水性过滤器过滤,浓缩,并在减压下干燥。获得25mg的n-[2-(2-羟乙基)-6-(羟甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(实施例7)。uplc-ms(方法a2):rt=0.87minms(esipos):m/z=381(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=3.87(q,2h),4.44(t,2h),4.69(d,2h),4.98(t,1h),5.70-5.81(m,1h),7.57(s,1h),8.11-8.23(m,1h),8.31-8.42(m,2h),8.43-8.49(m,1h),8.51(s,1h),11.20(s,1h)。实施例8n-[6-(2-羟基丙-2-基)-2-(氧杂环丁烷-3-基甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将50mg(0.12mmol)的2-(氧杂环丁烷-3-基甲基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-1)溶于500μl的thf中,并与576μl(0.58mmol)的1m的甲基溴化镁的thf溶液混合。将反应混合物在25℃下搅拌60min。随后,小心地加入20ml的饱和氯化铵水溶液,并将混合物浓缩。用乙酸乙酯萃取水相两次,合并有机相,用硫酸镁干燥,过滤,并浓缩。将残余物溶于2.0ml的dmso中,并通过制备型hplc纯化。将含有产物的馏分冷冻干燥。得到30mg的标题化合物。uplc-ms(方法a2):rt=1.03minms(esipos):m/z=435(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=1.62(s,6h),3.45-3.61(m,1h),4.48(t,2h),4.66(dd,2h),4.72(d,2h),5.94(s,1h),7.57(s,1h),8.16(d,1h),8.33-8.42(m,2h),8.42-8.47(m,1h),8.72(s,1h),12.36(s,1h)。实施例9n-[6-(羟甲基)-2-(氧杂环丁烷-3-基甲基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将75mg(0.17mmol)的2-(氧杂环丁烷-3-基甲基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-1)溶于1ml的thf/甲醇(1:1)混合物中,并加入8mg(0.21mmol)硼氢化钠。将混合物在25℃下搅拌60min。浓缩反应混合物,并将残余物与水混合。将悬浮液剧烈搅拌15min,抽滤出固体,用水洗涤两次,用二乙醚洗涤两次,并在减压下干燥。获得48mg的标题化合物。uplc-ms(方法a2):rt=0.94minms(esipos):m/z=407(m+h)+1hnmr(300mhz,dmso-d6):δ[ppm]=3.55(s,1h),4.48(t,2h),4.61-4.77(m,6h),7.57(s,1h),8.18(dd,1h),8.33-8.49(m,3h),8.51(s,1h),11.21(s,1h)。实施例10n-{6-(2-羟基丙-2-基)-2-[3-(甲基磺酰基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺将在5.0ml的dmf中的500mg(1.32mmol)的n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)、569mg的碳酸钾和114mg的碘化钾的混合物在室温下搅拌15min。加入414mg的1-溴-3-(甲基磺酰基)丙烷,并将混合物在室温下搅拌过夜。加入水,混合物用乙酸乙酯萃取两次,将萃取物用氯化钠溶液洗涤,并浓缩。残余物通过柱层析法纯化(二氯甲烷/甲醇梯度)。将产物部分与二乙醚一起搅拌、过滤并干燥。获得59mg的标题化合物。uplc-ms(方法a2):rt=1.02minms(esipos):m/z=485(m+h)+1h-nmr(300mhz,dmso-d6):δ[ppm]=1.63(s,6h),2.26-2.42(m,2h),2.99(s,3h),3.06-3.16(m,2h),4.55(t,2h),5.96(s,1h),7.60(s,1h),8.16(d,1h),8.33-8.48(m,3h),8.73(s,1h),12.37(s,1h)。实施例11n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺制备方法1首先将705mg(1.57mmol)的2-(3-羟基-3-甲基丁基)-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-4)加入到10ml的thf中,并在冰水冷却浴中冷却。加入2.6ml(5.0当量)3m的甲基溴化镁溶液(于二乙醚中),将混合物在用冰浴冷却的同时搅拌1h,并在室温下搅拌4.5h。另加入1当量的甲基溴化镁溶液,并将混合物在室温下搅拌20.5h。再加入1当量的甲基溴化镁溶液,并将混合物在室温下搅拌22h。将反应混合物与饱和氯化铵水溶液混合,搅拌,并用乙酸乙酯萃取三次。合并的有机相用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。获得790mg的残余物,将其通过制备型hplc纯化。获得234mg的标题化合物和164mg的产物部分,将所述产物部分与二乙醚一起搅拌。抽滤,然后干燥,这之后,又获得146mg的标题化合物。uplc-ms(方法a1):rt=1.10min(uv检测器:tic),分子量测定值450.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.14(s,6h),1.61(s,6h),1.99-2.08(m,2h),4.42-4.55(m,3h),5.93(s,1h),7.56(s,1h),8.15(dd,1h),8.32-8.39(m,2h),8.41-8.47(m,1h),8.70(s,1h),12.34(s,1h)。制备方法2将在5mldmf中的500mg(1.37mmol)n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)、569mg碳酸钾和114mg碘化钾的混合物在室温下搅拌15min。加入344mg(1.5当量)的4-溴-2-甲基丁-2-醇,并将混合物加热至100℃,保持2h。另加入0.5当量的4-溴-2-甲基丁-2-醇,并将混合物在室温下搅拌16h。将混合物与水混合并用乙酸乙酯萃取两次,将合并的有机相用饱和氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。将残余物通过硅胶柱层析法(己烷/乙酸乙酯)纯化。获得100mg的产物部分,将所述产物部分与二乙醚一起搅拌。过滤固体并干燥。获得60mg的标题化合物。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.14(s,6h),1.61(s,6h),1.99-2.07(m,2h),4.43-4.52(m,3h)5.94(s,1h)7.57(s,1h)8.15(dd,1h)8.33-8.40(m,2h),8.42-8.48(m,1h),8.71(s,1h),12.35(s,1h)。实施例12n-{6-(2-羟基丙-2-基)-2-[2-(甲基磺酰基)乙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺将160mg(0.44mmol)n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)与182mg碳酸钠和36mg碘化钾一起悬浮于1.0ml的dmf中,并将混合物在室温下搅拌15min。然后,加入123mg的2-溴乙基甲基砜(0.66mmol),并将混合物在室温下搅拌过夜。加入水,混合物用乙酸乙酯萃取两次,将萃取物用饱和氯化钠水溶液洗涤,通过疏水性过滤器过滤,并浓缩。通过制备型hplc纯化残余物,获得20mg的标题化合物。uplc(方法a2):rt=1.01min;ms(esipos):m/z=471(m+h)+1hnmr(400mhz,dmso-d6):δ[ppm]=1.63(s,6h),2.90(s,3h),3.85(t,2h),4.86(t,2h),5.97(s,1h),7.59(s,1h),8.13-8.19(m,1h),8.37(s,1h),8.41-8.48(m,2h),8.74(s,1h),12.37(s,1h)。实施例136-(二氟甲基)-n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]吡啶-2-羧酰胺制备方法1将在2.5mldmf中的250mg6-(二氟甲基)-n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]吡啶-2-羧酰胺(中间体5-2的粗产物)、144mg碘化钾和239mg碳酸钾混合物在室温下搅拌15min。加入145mg(0.87mmol)4-溴-2-甲基丁-2-醇,将混合物在110℃下搅拌3h,另加入96mg4-溴-2-甲基丁-2-醇,并将混合物在110℃下搅拌4h。加入水,混合物用乙酸乙酯萃取两次,将萃取物用半饱和氯化钠水溶液洗涤,通过疏水性过滤器过滤,并浓缩。通过硅胶柱层析法(己烷/乙酸乙酯)进行纯化。获得61mg的标题化合物。uplc-ms(方法a1):rt=1.00min(uv检测器:tic),分子量测定值432.00。1h-nmr(300mhz,dmso-d6):δ[ppm]=1.14(s,6h),1.63(s,6h),1.97-2.08(m,2h),4.41-4.55(m,3h),5.99(s,1h),7.03(t,1h),7.56(s,1h),7.94–8.00(m,1h),8.24-8.38(m,3h),8.71(s,1h),12.49(s,1h)。制备方法2类似于实施例11(制备方法1)的制备,使3.00g的5-({[6-(二氟甲基)吡啶-2-基]羰基}氨基)-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体4-11)与3m的甲基溴化镁溶液(于二乙醚中)反应。粗产物在与二乙醚一起进行搅拌、过滤后,通过制备型hplc纯化,获得1.37g的标题化合物。实施例146-(二氟甲基)-n-{6-(2-羟基丙-2-基)-2-[2-(甲基磺酰基)乙基]-2h-吲唑-5-基}吡啶-2-羧酰胺将在2.5mldmf中的250mg6-(二氟甲基)-n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]吡啶-2-羧酰胺(中间体5-2的粗产物)、144mg碘化钾和239mg碳酸钾的混合物在室温下搅拌15min。加入162mg2-溴乙基甲基砜(0.87mmol),并将混合物在110℃下搅拌3h。加入水,混合物用乙酸乙酯萃取两次,并将萃取物用半饱和氯化钠水溶液洗涤,通过疏水性过滤器过滤,并浓缩。残余物通过制备型hplc纯化,并将产物部分通过硅胶柱层析法(己烷/乙酸乙酯)再次纯化。得到40mg的标题化合物。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.65(s,6h),2.90(s,3h),3.85(t,2h),4.85(t,2h),6.03(s,1h),7.04(t,1h),7.59(s,1h),7.98(d,1h),8.25-8.36(m,2h),8.43(s,1h),8.75(s,1h),12.52(s,1h)。实施例156-(二氟甲基)-n-[6-(2-羟基丙-2-基)-2-(3-羟丙基)-2h-吲唑-5-基]吡啶-2-羧酰胺阶段a:n-[2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(二氟甲基)吡啶-2-羧酰胺的制备将在2.5mldmf中的250mg6-(二氟甲基)-n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]吡啶-2-羧酰胺(中间体5-2)、48mg碘化钾和239mg碳酸钾的混合物在室温下搅拌15min。加入219mg(0.87mmol,1.5当量)的(3-溴丙氧基)(叔丁基)二甲基硅烷,并将混合物在110℃下搅拌3h。另加入1当量的(3-溴丙氧基)(叔丁基)二甲基硅烷,并将混合物在100℃下搅拌4h。加入水,用乙酸乙酯萃取混合物,并将萃取物用氯化钠水溶液洗涤,通过疏水性过滤器过滤,并浓缩。残余物通过柱层析法(己烷/乙酸乙酯)纯化。获得92mg的标题化合物。阶段b:类似于实施例6步骤b的制备,使92mgn-[2-(3-{[叔丁基(二甲基)甲硅烷基]氧基}丙基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-(二氟甲基)吡啶-2-羧酰胺与0.53ml1m的四丁基氟化铵的thf溶液反应1h。如实施例6中进行水后处理,并通过制备型hplc进行纯化,获得46mg的标题化合物。uplc-ms(方法a1):rt=0.92min(uv检测器:tic),分子量测定值404.00。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.64(s,6h),2.05(quin,2h),3.35-3.46(m,2h),4.45(t,2h),4.64(t,1h),5.99(s,1h),7.04(t,1h),7.57(s,1h),7.95–7.99(m,1h),8.25-8.36(m,3h),8.73(s,1h),12.50(s,1h)。实施例16n-[6-(2-羟基丙-2-基)-2-(4,4,4-三氟丁基)-2h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺将210mg(0.58mmol)的n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)于3mldmf中的混合物与0.11ml(0.87mmol)的1,1,1-三氟-4-碘代丁烷和239mg的碳酸钾混合,并将混合物在80℃下搅拌6h。在加入水后,将混合物用乙酸乙酯萃取三次,并用饱和氯化钠溶液洗涤合并的有机相,通过疏水性过滤器过滤,并浓缩。粗产物通过制备型hplc纯化。获得19mg的标题化合物。uplc-ms(方法a1):rt=1.27min(uv检测器:tic),分子量测定值474.15。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.62(s,6h),2.10-2.33(m),4.49(t,2h),5.94(s,1h),7.59(s,1h),8.13-8.18(m,1h),8.32-8.41(m,2h),8.41-8.47(m,1h),8.72(s,1h),12.35(s,1h)。实施例17n-{6-(2-羟基丙-2-基)-2-[3-(三氟甲氧基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺首先将150mg(0.33mmol)n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)加入2mlthf中。加入58mg(0.40mmol)的3-(三氟甲氧基)丙-1-醇、131mg三苯基膦和71μl偶氮二甲酸二异丙酯(diad,cas2446-83-5),并将混合物在室温下搅拌19h。加入0.83ml的氢氧化钠溶液(2m),并将混合物在40℃下搅拌5h。用水稀释混合物,并用乙酸乙酯萃取三次,将合并的有机相浓缩,并通过制备型hplc纯化。获得16mg粗产物形式的标题化合物。uplc-ms(方法a2):rt=1.26min(uv检测器:tic),分子量测定值490.14。1h-nmr(400mhz,dmso-d6,选定的信号):δ[ppm]=1.61(s,6h),1.84(d,1h),2.32(quint.,2h),4.08(t,2h),4.51(t,2h),7.58(s,1h),8.15(d,1h),8.31–8.39(m,2h),8.44(d,1h),8.72(s,1h),12.35(s,1h)。实施例18n-{6-(2-羟基丙-2-基)-2-[3-(2,2,2-三氟乙氧基)丙基]-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺类似于实施例11的制备(制备方法1),使在3mlthf中的52mg(0.10mmol)的2-[3-(2,2,2-三氟乙氧基)丙基]-5-({[6-(三氟甲基)吡啶-2-基]羰基}氨基)-2h-吲唑-6-羧酸甲酯(中间体4-10)与2×171μl的3m的溴化镁的二乙醚溶液反应。通过制备型hplc纯化,获得12mg的标题化合物uplc-ms(方法a1):rt=1.25min(uv检测器:tic),分子量测定值504.16。1h-nmr(500mhz,dmso-d6):δ[ppm]=1.63(s,6h),2.20(quin,2h),3.58(t,2h),4.05(q,2h),4.47(t,2h),5.94(s,1h),7.58(s,1h),8.15(dd,1h),8.32(s,1h),8.36(t,1h),8.45(d,1h),8.73(s,1h),12.36(s,1h)。实施例195-氟-n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-甲基吡啶-2-羧酰胺首先将228mg(0.31mmol)的5-{[(5-氟-6-甲基吡啶-2-基)羰基]氨基}-2-(3-羟基-3-甲基丁基)-2h-吲唑-6-羧酸甲酯(中间体4-8)加入到4.5mlthf中,并用冰冷却浴冷却。加入0.63ml的3m的甲基溴化镁溶液(于二乙醚中),并将混合物在用冰浴冷却的同时搅拌2h,并在室温下搅拌21h。将反应混合物与饱和氯化铵水溶液混合,并用乙酸乙酯萃取三次。将合并的有机相浓缩。残余物通过制备型hplc纯化。获得82mg的标题化合物。uplc-ms(方法a2):rt=1.03min(uv检测器:tic),分子量测定值414.21。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.13(s,6h),1.63(s,6h),1.99-2.05(m,2h),2.55-2.59(m,3h),4.42-4.50(m,3h),5.95(s,1h),7.54(s,1h),7.83(t,1h),8.05(dd,1h),8.31(s,1h),8.68(s,1h),12.33(s,1h)。实施例20n-[2-(3-羟基-3-甲基丁基)-6-(2-羟基丙-2-基)-2h-吲唑-5-基]-6-甲基吡啶-2-羧酰胺首先将278mg(0.48mmol)2-(3-羟基-3-甲基丁基)-5-{[(6-甲基吡啶-2-基)羰基]氨基}-2h-吲唑-6-羧酸甲酯(中间体4-9)加入到5.0mlthf中,并用冰冷浴冷却。加入0.97ml3m的甲基溴化镁溶液(于二乙醚中),将混合物在用冰浴冷却的同时搅拌2h,并在室温下搅拌20.5h。另加入0.48ml3m的甲基溴化镁溶液,并将混合物在室温下搅拌67h。将混合物与饱和氯化铵水溶液混合,并用乙酸乙酯萃取三次,萃取物用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。残余物通过制备型hplc纯化。获得111mg的标题化合物。uplc-ms(方法a2):rt=0.97min(uv检测器:tic),分子量测定值396.22。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.15(s,6h),1.64(s,6h),2.00-2.08(m,2h),2.61(s,3h),4.41-4.59(m,3h),5.92(s,1h),7.50(dd,1h),7.56(s,1h),7.90-7.99(m,2h),8.33(s,1h),8.70(s,1h),12.39(s,1h)。实施例216-(2-羟基丙-2-基)-n-[6-(2-羟基丙-2-基)-2-(4,4,4-三氟丁基)-2h-吲唑-5-基]吡啶-2-羧酰胺将72mg(0.16mmol)的5-({[6-(2-羟基丙-2-基)吡啶-2-基]羰基}氨基)-2-(4,4,4-三氟丁基)-2h-吲唑-6-羧酸甲酯(中间体4-7)于10ml的thf中的溶液在冰/水冷却浴中冷却。加入0.26ml的3m的甲基溴化镁的二乙醚溶液,并将混合物搅拌2h,然后在室温下搅拌20h。另加入1当量3m的甲基溴化镁溶液,并将混合物在室温下搅拌24h。加入饱和氯化铵水溶液,混合物用乙酸乙酯萃取三次,将萃取液用氯化钠溶液洗涤,并浓缩。通过制备型hplc获得22mg(理论值的31%)的标题化合物。uplc-ms(方法a2):rt=1.15min(uv检测器:tic),分子量测定值464.20。1h-nmr(400mhz,dmso-d6):δ[ppm]=1.56(s,6h),1.64(s,6h),2.07-2.34(m,4h),4.49(t,2h),5.32(s,1h),6.05(s,1h),7.60(s,1h),7.87(dd,1h),7.99-8.05(m,2h),8.35(s,1h),8.79(s,1h),12.45(s,1h)。实施例22n-{2-[2-(1-羟基环丙基)乙基]-6-(2-羟基丙-2-基)-2h-吲唑-5-基}-6-(三氟甲基)吡啶-2-羧酰胺首先将250mg(0.69mmol)的n-[6-(2-羟基丙-2-基)-1h-吲唑-5-基]-6-(三氟甲基)吡啶-2-羧酰胺(中间体5-1)加入到5mldmso中。加入159mg(0.96mmol)1-(2-溴乙基)环丙醇、285mg碳酸钾和171mg碘化钾,并将混合物在100℃下搅拌5h。加入水,并将混合物用乙酸乙酯萃取三次。将合并的有机相用氯化钠溶液洗涤,通过疏水性过滤器过滤,并浓缩。残余物通过制备型hplc纯化(柱:watersxbridgec185μ100×30mm,洗脱液a:水+0.1体积%的甲酸(99%),洗脱液b:乙腈)。冷冻干燥获得45mg的标题化合物。1h-nmr(500mhz,dmso-d6):δ[ppm]=0.18-0.22(m,2h),0.48-0.52(m,2h),1.62(s,6h),2.08(t,2h),4.54-4.60(m,2h),5.36(s,1h),5.96(s,1h),7.57(s,1h),8.16(dd,1h),8.34-8.39(m,2h),8.45(d,1h),8.72(s,1h),12.36(s,1h)。生理功效评估irak4激酶试验本发明物质对irak4-抑制的活性在下文中记载的irak4tr-fret试验(tr-fret=时间分辨荧光共振能量转移)中测定。将来自n-末端的gst(谷胱甘肽-s-转移酶)和人irak4重组融合蛋白用作酶,所述重组融合蛋白在杆状病毒感染的昆虫细胞(hi5,bti-tn-5b1-4,购自invitrogen的细胞系,目录号b855-02)中表达,并通过亲和色谱法纯化。用于激酶反应的底物为生物素化的肽生物素-ahx-kkarfsrfagsspsqasfaepg(酰胺形式的c末端),其可购自例如biosyntangmbh(berlin-buch)。为进行试验,由试验物质的2mm的dmso溶液制备20μm至0.073nm范围内的11个不同的浓度。将50nl的相应的溶液吸移到黑色的低容量384-孔微量滴定板(greinerbio-one,frickenhausen,德国)中,加入2μlirak4于试验缓冲液[50mm的ph为7.5的hepes,5mm的mgcl2,1.0mm的二硫苏糖醇,30μm的活化的原钒酸钠,0.1%(w/v)的牛γ-球蛋白(bgg),0.04%(v/v)的诺乃洗涤剂(nonidet)-p40(sigma)]中的溶液,并将混合物温育15min以使所述物质在进行激酶反应之前预先结合到酶上。然后通过加入3μl的三磷酸腺苷(atp,1.67mm=在5μl的试验体积中的最终浓度:1mm)和肽底物(0.83μm=在5μl的试验体积中的最终浓度:0.5μm)于试验缓冲液的溶液启动激酶反应,并将所得混合物在22℃下温育45min的反应时间。irak4的浓度调节至酶各自的活性,并设置使得试验在线性范围内进行。通常浓度在约0.2nm的量级上。通过加入5μl的tr-fret检测试剂[0.1μm的抗生蛋白链菌素-xl665(cisbiobioassays;法国,目录号610saxlg)]和1.5nm的抗磷酸丝氨酸抗体[merckmillipore,“stk抗体”,目录号35-002]以及0.6nm的lanceeu-w1024标记的抗小鼠igg抗体(perkin-elmer,产品号ad0077;或者可使用来自cisbiobioassays的铽穴合物标记的抗小鼠igg抗体)于edta水溶液(100mm的edta,0.4%(w/v)的牛血清白蛋白[bsa]于25mm的ph为7.5的hepes中)中的溶液来终止反应。将所得混合物在22℃下温育1h以形成生物素化的磷酸化底物和检测试剂的复合物。然后通过测定从铕螯合物标记的抗小鼠igg抗体转移到链霉抗生素蛋白-xl665的共振能量来评估磷酸化底物的量。为此,在tr-fret测量仪器如rubystar(bmglabtechnologies,offenburg,德国)或viewlux(perkin-elmer)中在350nm激发后,测量在620nm和665nm的荧光发射。将在665nm和622nm处的发射比作为磷酸化底物的量的量度。将数据归一化(没有试验物质的酶反应=0%抑制;所有其他试验组分但没有酶=100%抑制)。通常,将试验物质以20μm至0.073nm范围内的11个不同的浓度(20μm、5.7μm、1.6μm、0.47μm、0.13μm、38nm、11nm、3.1nm、0.89nm、0.25nm和0.073nm)于相同的微量滴定板上测试。稀释系列在试验之前通过系列稀释制备(2mm至7.3nm,于100%的dmso中)。通过4-参数拟合计算ic50值。表1:在irak4激酶试验中实施例化合物的ic50值在如上所述的irak4tr-fret试验中同样测定了本发明通式(iii)的物质对于irak4的抑制活性。以下以举例的方式提及:化合物中间体4-2,ic50=21.7nm;中间体4-3,ic50=13.0nm;和中间体4-4,ic50=6.2nm。thp-1细胞中的tnf-α分泌该试验适用于试验物质抑制thp-1细胞(人单核细胞急性白血病细胞系)中tnf-α(肿瘤坏死因子α)分泌的能力。tnf-α是参与炎症性过程的细胞因子。在该试验中,tnf-α分泌通过细菌脂多糖(lps)温育而引发。将thp-1细胞保存于连续悬浮细胞培养基[含有l-glutamax的rpmi1460培养基(gibco,目录号61870-044),补充有10%的胎牛血清(fcs)(invitrogen,目录号10082-147)、1%的青霉素/链霉素(gibcobrl,目录号15140-114)]中,并且不应超过1×106个细胞/ml的细胞浓度。所述试验在细胞培养基(含有l-glutamax的rpmi1460培养基,补充有10%的fcs)中进行。在每种情况下,将每孔2-2.5μl的细胞悬浮液(对应4000个细胞)分配到384-孔测试板(greiner,目录号784076)中,其中在每个孔中,已将40-50nl的物质溶解于100%的dmso中。对于每种物质,溶解都通过使用在20μm至0.073nm范围内的10个不同的浓度来完成。将细胞在室温下温育15min。然后将2-2.5μl的溶于细胞培养基中的0.1μg/ml的lps(sigma,大肠杆菌(escherichiacoli)055:b5,目录号l5418)(最终浓度0.05μg/ml)分配到各孔中。作为中性对照,细胞用0.05μg/ml的lps和1%的dmso处理,作为抑制剂对照,仅用1%的dmso处理。将板以80g离心30s,并在37℃、5%co2和95%大气湿度下温育17h。tnf-α的量使用tnf-αhtrf检测试剂盒(detectionkit)(cisbio,目录号62tnfpeb/c)测定。为此,在各情况下,加入2μl的检测溶液——其由抗-tnf-α-xl665缀合物和抗-tnf-α-穴合物缀合物组成,根据制造商的说明书溶于重建缓冲液中——用于htrf(均匀时间分辨荧光)测试。加入后,将混合物在室温下温育3h或在4℃下温育过夜。然后使用支持htrf的测量仪器如bmgpherastar读取620/665nm处的信号。物质的活性表示为以百分比计的中性对照和抑制剂对照之间的比值。使用4-参数拟合计算ic50值。表2:实施例化合物关于thp-1细胞中tnf-α分泌的ic50值体外lps(脂多糖)-诱导的人pbmc(外周血单核细胞)中细胞因子的生成考察了本发明通式(i)的化合物对于人pbmc中诱导性细胞因子生成的功效。本文中,细胞因子产生由lps诱导—一种tlr4配体,其导致irak4-介导的信号途径的激活。人pbmc由人抗凝全血获得。为此目的,首先将15ml的ficoll-paque(biochrom,目录号l6115)吸移到leucosep管中,并加入20ml的人血。将该血在室温下800g离心15min后,移除并弃去包含血小板的血浆。将pbmc转移到离心管中,并补充pbs(磷酸盐缓冲盐水)(gibco,目录号14190)。将细胞悬浮液在室温下250g离心10min并弃去上清液。将pbmc重新悬浮于完全培养基(rpmi1640,不含l-谷氨酰胺(paa,目录号e15-039),10%的fcs;50u/ml青霉素,50μg/ml链霉素(paa,目录号p11-010)和1%l-谷氨酰胺(sigma,目录号g7513))中。试验也是在完全培养基中进行。将pbmc以2.5×105个细胞/孔的细胞密度接种到96-孔板上。将本发明化合物进行一系列稀释,稀释在等容的100%dmso中,并以10μm至3nm范围内的8个不同浓度应用于试验中,使得最终的dmso浓度为0.4%dmso。然后在实际刺激之前,将细胞与其预温育30min。为诱导细胞因子分泌,用0.1μg/ml的lps(sigma,大肠杆菌(escherichiacoli)0128:b12,目录号l2887)刺激细胞24小时。细胞生存力根据制造商的说明书使用celltiter-glo发光测定法(promega,目录号g7571(g755/g756a))进行测定。细胞培养物上清液中分泌的tnf-α的量根据制造商的说明书使用humanproinflammatory9-plextissueculturekit(msd,目录号k15007b)测定。例如,实施例化合物11和实施例化合物12的活性≤1μm。体外tlr-4/tlr-7诱导的人树突细胞(dc)的白细胞介素(il)-23的分泌在人dc中考察本发明通式(i)的化合物对促炎细胞因子il-23的诱导生成的功效,所述促炎细胞因子il-23对th-17细胞的生成起重要作用。据报道,th-17细胞在疾病如类风湿性关节炎、牛皮癣关节炎、别赫捷列夫病(强直性脊柱炎(ankylosingspondylitis))或多发性硬化症的发病机理中起着关键作用(lubberts,nat.rev.rheumatol.,2015;marinoni等人,auto.immun.highlights,2014;isailovic等人,j.autoimmun.,2015;staschke等人,jimmunol.,2009)。为检测本发明化合物对il-23生成的影响,使人原代单核细胞(使用磁分离法[miltenyibiotech,单核细胞分离试剂盒(monocyteisolationkit),目录号130-091-153]从人pbmc中分离,并在完全培养基(vle(非常低的内毒素)rpmi1640[biochromag,目录号fg1415]、10%的胎牛血清(fbs)[gibco,目录号10493-106];50μm的β-巯基乙醇([gibco,目录号31350]、50u/ml的青霉素和链霉素[gibco,目录号15140-114])中加入生长因子(重组人gm-csf[peprotech,目录号300-03]和il-4[peprotech,目录号200-04]))在培养基中在6天内分化为dc。在收集dc后,将其重新悬浮于完全培养基中,并以2×105个细胞/孔的细胞密度接种到96-孔板(costar,目录号3599)中。将本发明化合物进行一系列稀释,稀释在等容的100%dmso中,并以10μm至1nm范围内的9个不同浓度用于试验中。此处确保,对于所使用的9个浓度中的每一个,存在的dmso浓度总为0.1%。将dc与本发明化合物一起预温育30min。其后,通过加入10ng/ml的lps(sigma,大肠杆菌(escherichiacoli)血清型0127:b8,目录号l3129)(tlr4配体)和2.5μg/ml的tlr-7/8配体r848(invivogen,目录号tlrl-r848-5)(二者都可激活irak4介导的信号途径),在培养箱(37℃,95%的rh,5%的co2)中刺激dc以产生il-23,进行24小时。在24小时的温育时间后,收集上清液,并根据制造商的说明书,使用市售可得的hil-23elisa(ebiosciences,目录号88-7237-88)进行分析。以实施例化合物12为例,对人dc中il-23的抑制的结果示于图1中。体外tlr-7/8-或tlr-9诱导的人浆细胞样树突细胞(pdc)的ifnα的生成借助该试验,可研究本发明通式(i)的化合物对人pdc中的ifnα(干扰素α)(系统性红斑狼疮发病机理中的关键细胞因子)的生成的影响(mathian等人,arthritisrheum,2009;crowm.k.,rheumdisclinnam,2010)。为此目的,如上所述,从全血中分离人pbmc,并使用市售可得的细胞分离试剂盒(miltenyibiotech,plasmacytoiddendriticcellisolationkitii,目录号130-097-415)从中分离出浆细胞样dc(pdc)。将获得的pdc重新悬浮于完全培养基(rpmi1640+glutamax[gibco,目录号61870-010],补充10%的fbs[gibco,目录号10493-106]和50u的青霉素/链霉素[gibco,目录号15140-114])中,并以5×104个细胞/孔的细胞密度接种于96-孔的微量滴定板(costar,目录号3599)中。将本发明化合物在等容的100%dmso中进行一系列稀释,并以10μm至1nm范围内的9个不同的浓度用于测试中。对于9个测试浓度中的每一个,确保存在的dmso浓度总是为0.1%。将pdc与本发明化合物一起预温育30分钟。用tlr7/8配体(咪喹莫特(imiquimod),r837,invivogen,目录号tlrl-imq)或用tlr-9配体(cpg-a,odn2216,invivogen,目录号tlrl-2216-1)刺激pdc,这会导致irak4介导的信号途径的激活。在温育24小时后,除去细胞培养物上清液,并使用市售可得的人ifnαelisa(ifnalphamulti-subtypeelisakit,pblassayscience,目录号41105-1)进行分析。以实施例化合物12为例,对人浆细胞样dc中ifnα的抑制的结果示于图2中。tlr-介导的炎症的体内模型在体内tlr-介导的炎症模型中考察本发明通式(i)的化合物的体内功效。由于使用了lps-介导的炎症模型,该机理模型特别显示了本发明化合物对于tlr4-介导的疾病的潜在功效。在该模型中,将雌性balb/c小鼠(约8周大;charlesriver实验室,德国)分组,每组5只动物。对照组用其中溶解有所述物质的媒介物(vehicle)(物质媒介物)和其中溶解有lps的媒介物进行治疗。对物质治疗组和阳性对照组以0.2mglps/kg体重(sigma,目录号l4391)(脂多糖来自e.coli0111:b4)经腹膜内(i.p.)给药。此外,用上述物质媒介物治疗阳性对照组。在通过给予lps诱导炎症前16小时,口服给药所述物质。为考察本发明的化合物对炎症的功效,在1.5小时后从动物中采取血样。根据制造商的说明书,使用mouseproinflammatory7-plextissueculturekit(msd,目录号k15012b)测定血浆中特定细胞因子的浓度。irak4抑制剂在tlr-介导的炎症模型中是有效的。图3示显示了血浆中tnf-α的量,与lps-诱导的浓度相比,其通过给药实施例化合物11以剂量依赖性方式降低。il-1β-介导的炎症的体内模型为评估本发明通式(i)的化合物在il-1β-介导的疾病中的潜在功效,对雌性balb/c小鼠(约8周大;charlesriver实验室,德国)经腹膜内给予il-1β,并考察本发明化合物对于il-1β-介导的细胞因子分泌的功效。每组有5只动物。对照组用能够用于溶解所述物质和il-1β的媒介物治疗。对物质治疗组和阳性对照组分别经腹膜内给予90μgil-1β/kg体重(r&d,目录号401-ml/cf)。在给予il-1β之前6小时,在阳性对照组中给予所述物质或其媒介物。在给予il-1β之后2小时,根据制造商的说明书,使用mouseproinflammatory7-plextissueculturekit(msd,目录号k15012b)测定从血液分离的血浆中的tnf-α。给予il-1β导致tnf-α血浆浓度升高,其通过用实施例化合物11和12治疗而抑制。这由图4说明。体内佐剂诱导的关节炎模型为测定本发明通式(i)的化合物的抗炎症活性,在关节炎模型中考察其体内功效。为此目的,在第0天,将各雄性lewis大鼠(约100-125g,charlesriverlaboratories,germany)向尾基部皮下给予100μl的完全弗氏佐剂(freund'sadjuvant)(cfa)溶液(结合分枝杆菌(m.tuberculosis)h37ra[difolab,目录号-231141],溶于不完全弗式佐剂[difcolab,目录号-263910]中)。在每组中有n=8只大鼠。健康对照组和患病对照组都纳入研究中。各对照组仅用试验物质的媒介物口服(p.o.)治疗。以预防性的方式,即从第0天开始,用不同剂量的试验物质通过口服给药进行治疗。在第0天,根据疾病活动评分(基于点系统的关节炎严重程度分级)另外确定动物的起始状况。本文中,对于两个后爪一起的包括关节肿胀在内的红斑,根据关节炎症的程度,给予0至4点(0=无;1=轻度;2=中度;3=明显;4=严重)。为测定化合物的抗炎功效,从第8天开始,当动物最先出现关节炎迹象时,借助疾病活性评分对动物的疾病活动性进行评分,随后每周进行3次,直至结束(第20天)。使用单因素方差分析(anova)进行统计分析,并借助多重比较分析(dunnett's检验)与对照组进行比较。在大鼠中皮下给予cfa导致大鼠具有明显关节炎症的急性关节炎。通过用实施例化合物11治疗来抑制这种诱导性的关节炎。这由图5说明。小鼠体内胶原抗体诱导的关节炎模型在另一鼠类关节炎模型中考察本发明通式(i)的化合物的抗炎功效。为此目的,在第0天,将每只雌性balb/c小鼠(约9周大,charlesriverlaboratories,kingston,canada)向尾静脉中静脉内注射200μl胶原抗体混合剂(10mg/ml;arthritomab,mdbioproducts)(研究中包括的健康对照组除外)。然后在第6天,使这些小鼠再分别接受腹膜内注射200μllps。每组有n=10只小鼠。健康对照组和患病对照组都纳入研究中。各对照组仅用试验物质的媒介物进行口服治疗。以预防性的方式,即从第0天开始,使用不同剂量的试验物质通过口服给药进行治疗。在实验过程中,基于用于疾病活动评分的点判定系统,对所有四只爪的患病程度进行评分。在该点判定中,对于健康的爪判定为没有点,而对于从脚趾通过跖关节至踝关节发生的特定程度的关节发炎,分别判定为1点[轻度发炎,例如脚趾发炎]至4分[延伸到整个爪子的严重发炎],说明如下:●0=正常●1=限于跗节或踝关节或脚趾的红斑和轻度肿胀●2=从踝关节延伸至跖骨的红斑和轻度肿胀(2阶段)●3=从踝关节延伸至远至跖骨关节的红斑和中度肿胀●4=涵盖跖骨、足和趾的红斑和严重肿胀对于该参数,在开始实验的前一天(第-1天)事先确定起始状况,并且随后从第8天往后,对该疾病活动评分每周评分三次。使用单因素方差分析(anova)进行统计分析,并借助多重比较分析(dunnett's检验)与对照组进行比较。在小鼠内经静脉给予胶原抗体混合剂,包括随后腹膜内给予lps,导致具有明显的关节炎症的急性关节炎。通过用实施例化合物12治疗,该诱导性关节炎被抑制。这由图6说明。体内nash小鼠模型为了实验诱导nash,在45只雄性2天大的c57bl/6小鼠中分别皮下注射200μg的链脲霉素(stz;sigma-aldrich,usa)。从4周龄开始,对这些动物随意喂食高脂肪饮食(hfd;57kcal%脂肪,购自clea,japan的#hfd32)。在6周龄时,将动物随机分为3组(每组15只动物)。其中一组不接受任何治疗,但另外2组在4周内每天口服给予媒介物或试验物质。在4周的治疗后,将所有动物在麻醉下无痛处死,移去肝脏并固定在布安溶液(bouin'ssolution)中,以用于组织学研究(h.denk,"fixierunghistologischer"[fixingofhistologicalpreparations],in:(编辑):"romeismikroskopischetechnik"[romei'smicroscopytechniques],urban&schwarzenberg,munich-vienna-baltimore1989,第17版,第97页,isbn3-541-11227-1)。其后,将肝脏样品包埋于石蜡中,并制备5μm厚的石蜡切片。将各肝脏的组织切片染色,a)用苏木精-伊红(hc)染色,用于测定nafld活性评分(nas),b)用苦天狼星红(picro-siriusred)染色(waldeck,germany),用于测定肝纤维化。基于d.e.kleiner等人,hepatology41(2005),1313-1321(表1)所推荐的标准,在苏木精-伊红切片中测定nafld活性评分。对于纤维化区域的组织学定量,在200倍的显微镜放大下,对各切片拍摄5张数码照片(dfc280;leica,germany),并使用imagejsoftware(国立卫生研究院,usa)测定纤维化百分比。体内db/db小鼠模型使用30只8周龄的雄性db/db小鼠。该模型是广泛接受的肥胖、胰岛素抵抗和2型糖尿病模型(aileenjfking;theuseofanimalmodelsindiabetesresearch;britishjournalofpharmacology166(2012),877–894)。在实验期间,使动物接受标准饮食(rm1(e)801492,sds),并自由饮用自来水。将动物随机分为3组(每组10只),并在6周内用试验物质口服治疗。在研究期间,在不同时间点(在治疗开始前、治疗开始之后3周和治疗结束之前2天)从动物中取血,以测定胰岛素敏感性参数(例如hba1c、葡萄糖含量、胰岛素含量)。此外,作为用于确定胰岛素敏感性的参数的ogtt(口服葡萄糖耐量试验)是在治疗开始之前1天和治疗结束之后2天进行。此外,计算homa-ir指数(空腹胰岛素水平(mu/l)*空腹血糖水平(mmol/l)/22.5)。体内b细胞淋巴瘤相关的异种移植模型在鼠类异种移植模型中研究本发明通式(i)的化合物的抗肿瘤活性。为此目的,对雌性c.b-17scid小鼠皮下植入人b细胞淋巴瘤细胞系,例如tmd-8。在平均肿瘤大小为20-30mm2的情况下,开始用本发明化合物口服单药治疗,或将本发明化合物与标准疗法相结合,每种都为口服给药。然而,动物事先是随机的。一旦未经治疗的对照组具有大的肿瘤,则立即结束治疗。肿瘤大小和体重每周测量三次。体重减轻是治疗-相关毒性的量度(>10%=临界,停止治疗直到恢复,>20%=中毒,终止)。通过电子测径规检测肿瘤面积[长度(mm)×宽度(mm)]。在研究结束时,还测定肿瘤重量。抗肿瘤功效定义为治疗组与对照组的肿瘤重量之比(t/c)[治疗组在第x天的肿瘤重量/对照组在第x天的肿瘤重量]或治疗组与对照组的肿瘤面积之比[治疗组在第x天的肿瘤面积/对照组在第x天的肿瘤面积]。t/c大于0.5的化合物被定义为有活性的(有效的)。使用单因素anova进行统计分析,并通过与对照组进行比较逐对对比分析(dunnett's检验)。犬irak4激酶试验本发明化合物对犬irak4的irak-4抑制活性在下文中所述的irak4tr-fret试验(tr-fret=时间分辨荧光共振能量转移)中测定。来自n-末端的his(多组氨酸)和犬irak4的重组融合蛋白,其在杆状病毒感染的昆虫细胞(hi5,bti-tn-5b1-4,购自invitrogen的细胞系,目录号b855-02)中表达,并通过亲和色谱法纯化,被用作酶。用于激酶反应的底物为生物素化的肽生物素-ahx-kkarfsrfagsspsqasfaepg(酰胺形式的c末端),其可从例如由biosyntangmbh(berlin-buch)购得。为进行试验,由2mm的试验物质的dmso溶液制备在20μm至0.073nm范围内的11种不同的浓度。将50nl的相应的溶液吸移到黑色的低容量384-孔微量滴定板(greinerbio-one,frickenhausen,德国)中,加入2μl的irak4于试验缓冲液[50mm的ph为7.5的hepes,5mm的mgcl2,1.0mm的二硫苏糖醇,30μm的活化的原钒酸钠,0.1%(w/v)的牛γ-球蛋白(bgg),0.04%(v/v)的诺乃洗涤剂-p40(sigma)]中的溶液,并将混合物温育15min以使所述物质在进行激酶反应之前预先结合到酶上。然后通过加入3μl的三磷酸腺苷(atp,1.67mm=在5μl的试验体积中的最终浓度:1mm)和肽底物(0.83μm=在5μl的试验体积中的最终浓度:0.5μm)于试验缓冲液中的溶液开始激酶反应,并将所得混合物在22℃下温育45min的反应时间。将irak4的浓度调节至酶的各自的活性,并设置使得试验在线性范围内进行。通常浓度在约0.1nm的量级。通过加入5μl的tr-fret检测试剂[0.1μm的抗生蛋白链菌素-xl665(cisbiobioassays;法国,目录号610saxlg)]、1.5nm抗磷酸丝氨酸抗体[merckmillipore,“stk抗体”,目录号35-002]和0.6nmlanceeu-w1024标记的抗小鼠igg抗体(perkin-elmer,产品号ad0071;或者可使用来自cisbiobioassays的铽穴状化合物标记的抗小鼠igg抗体)于edta水溶液(100mm的edta,0.4%(w/v)的牛血清白蛋白[bsa]于25mm的ph为7.5的hepes中)中的溶液来终止反应。将所得混合物在22℃下温育1h以形成生物素化的磷酸化底物和检测试剂的复合物。然后通过测定从铕螯合物标记的抗小鼠igg抗体转移到链霉抗生素蛋白-xl665的共振能量来评估磷酸化底物的量。为此,在tr-fret测量仪器如rubystar(bmglabtechnologies,offenburg,德国)或viewlux(perkin-elmer)中在350nm激发后,测量在620nm和665nm的荧光发射。将在665nm和622nm处的发射比作为磷酸化底物的量的量度。将数据归一化(没有试验物质的酶反应=0%抑制;所有其他试验组分但没有酶=100%抑制)。通常,将试验物质以20μm至0.073nm范围内的11个不同的浓度(20μm、5.7μm、1.6μm、0.47μm、0.13μm、38nm、11nm、3.1nm、0.89nm、0.25nm和0.073nm)于相同的微量滴定板上进行测试。稀释系列在试验之前通过系列稀释制备(2mm至7.3nm于100%的dmso中)。通过4-参数拟合计算ic50值。表3:在irak4犬激酶试验中的实施例化合物的两次实验的ic50值体外脂多糖(lps)诱导的通过犬外周血单核细胞(pbmc)的细胞因子的生成考察了本发明通式(i)的化合物对犬pbmc中诱导性细胞因子生成的功效。本文中细胞因子生成由lps诱导—一种tlr4配体,其导致irak4-介导的信号途径的激活。犬pbmc由狗抗凝全血获得。为此目的,通过将15ml狗血在4℃,400g离心15min来制备富含犬白细胞的血浆,随后收集,然后将犬pbmc血沉棕黄层悬浮于血浆中。将7(7)ml的ficoll-paqueplus(fischerscientific,目录号11778538)吸移到离心管中,然后使7ml的富含犬白细胞的血浆在ficoll-paqueplus顶部分层。在4℃下以将该管400g离心20min后,从犬血浆和ficoll-paqueplus的界面处收集犬pbmc。将pbmc转移到新的离心管中,并补充不含ca2+/mg2+(sigma-aldrich,目录号h9394)的hanks’平衡盐溶液1x(hbss)。将细胞悬浮液在4℃下以400g离心5min,然后弃去上清液。然后将细胞沉淀重新悬浮于0.2%的低渗盐水中,以溶解任何残余的红细胞。在30秒后,使细胞悬浮液等渗,并在4℃下以400g离心5min。然后将细胞沉淀重新悬浮于不含ca2+/mg2+的hbss中进行最后一次洗涤,并在4℃下以400g离心5min。然后,将pbmc重新悬浮于完全培养基(rpmi1640glutamax(sigma-aldrich,目录号r0883),10%的fcs;50u的青霉素,50μg/ml的链霉素(sigma-aldrich,目录号p4333))中。试验也在完全培养基中进行。将pbmc以2.5×105个细胞/孔的细胞密度接种到96-孔板上。将本发明化合物溶于dmso中,并在完全培养基中进行一系列稀释。化合物实施例在试验中以在3nm至10μm范围内的8个不同的浓度使用,使得最终的dmso浓度为0.0003–0.4%。为诱导细胞因子分泌,用0.1μg/ml的lps(sigma-aldrich,大肠杆菌(escherichiacoli)0111:b4,目录号l3024)刺激细胞,保持24小时。使用0.2%的台盼蓝(sigma-aldrich,目录号t8154)测定细胞生存力。根据制造商的说明书,使用犬tnfαduosetelisa(r&dsystems,目录号dy1507)测定细胞培养物上清液中所分泌的tnf-α的量。例如,实施例化合物12抑制通过lps刺激犬pbmc的tnfα的生成。体外脂多糖(lps)诱导的通过牛外周血单核细胞(pbmc)的细胞因子的生成考察了本发明通式(i)的化合物对牛pbmc中诱导性细胞因子生成的功效。本文中细胞因子生成由lps诱导—一种tlr4配体,其导致irak4-介导的信号途径的激活。牛pbmc由牛抗凝全血获得。为此目的,通过将500ml牛血在室温(rt)下1000g离心20min来制备富含牛白细胞的血浆,随后收集,然后将牛pbmc血沉棕黄层悬浮于等体积的pbs/5mmedta(室温)中。将30(30)ml的ficoll-paqueplus(fischerscientific,目录号11778538)吸移到leucosep管中,然后使30ml的牛pbmc血沉棕黄层/pbs/edta混合物在ficoll-paqueplus顶部分层。在室温下以800g离心25min后,从牛血浆和ficoll-paqueplus的界面处收集牛pbmc。将pbmc转移到新的离心管中,并补充冷的pbs/5mmedta。将细胞悬浮液在4℃下以350g离心10min,并弃去上清液。然后将细胞沉淀重新悬浮于0.2%的低渗盐水中,以溶解任何残余的红细胞。在30秒后,使细胞悬浮液等渗,并在4℃下以500g离心5min。然后将细胞沉淀重新悬浮于完全培养基(dmemwithglutamax(thermofisher,目录号32430100),10%的马血清(30-2040tm),20μm的β-巯基乙醇(thermofisher目录号31350010[贮存液:50mm])中。试验也在完全培养基中进行。将pbmc以1×106个细胞/孔的细胞密度接种到24-孔板上。将本发明化合物溶于dmso中,并在完全培养基进行一系列稀释。化合物实施例在试验中以在0.003μm至10μm范围内的8个不同的浓度使用,使得最终的dmso浓度为0.5%。为诱导细胞因子分泌,用1μg/ml(图8)和0.1μg/ml(图9)的lps(lps来自e.colik12;invivogen#tlrl-eklps)刺激细胞24小时。使用türk溶液(merckmillipore#1092770100)测定细胞生存力。使用基于兔抗牛tnfα抗体(rabbitanti-bovinetnfαantibody)的elisa读出装置(read-out)测定暴露于lps的牛pbmc的细胞培养物上清液中所分泌的tnfα的量。elisa试验在384孔elisa板中进行,所述elisa板用在50mmph9.6的na2co3/nahco3缓冲液中的5μg/ml兔抗牛tnfα抗体(biorad,ahp2383)以10μl/孔包被并在4℃下过夜。在除去抗体并用50μl的清洗缓冲液(pbs,0.05%(v/v)tween20)将孔冲洗三次后,用40μl的封闭缓冲液(pbs,0.05%(v/v)tween20,1%(w/v)牛血清白蛋白)将孔在37℃下温育90min。其后,除去封闭缓冲液,并加入培养物上清液样品(20μl/孔)。在37℃下温育90min后,除去样品,并用50μl的清洗缓冲液将孔冲洗三次。将1μg/ml的在封闭缓冲液中的兔抗牛tnfα生物素缀合抗体(biorad,ahp2383b)加入到在37℃下温育60min的板(20μl/孔)中。在除去生物素化的抗体并用50μl的清洗缓冲液将孔冲洗三次后,在37℃下加入1:10.000稀释在封闭缓冲液中的extravidintm-碱性磷酸酶(sigma,e2636),20μl/孔,保持1h。在除去extravidintm-碱性磷酸酶并用50μl的清洗缓冲液将孔冲洗三次后,通过加入50μl/孔的显色缓冲液(于50mmph9.6的na2co3/nahco3,2mmmgcl2中的5mm的对硝基苯磷酸酯(pnpp))引发酶促反应/显色。记录在405nm波长下的光密度。对于动力学测量,数据点每隔5分钟记录一次,记录1小时,在2小时后进行终点测量。例如,实施例化合物12抑制通过lps刺激的牛pbmc的tnfα的生成。这由图8和图9说明。体外脂多糖(lps)诱导的通过猪外周血单核细胞(pbmc)的细胞因子的生成作为另一实施例,考察了本发明通式(i)的化合物对猪pbmc中诱导性细胞因子生成的功效。本文中细胞因子生成由lps诱导—一种tlr4配体,其导致irak4-介导的信号途径的激活。猪pbmc由猪抗凝全血获得。为此目的,通过将36ml猪血在室温(rt)下1000g离心20min来制得富含猪白细胞的血浆,随后收集,然后将猪pbmc血沉棕黄层悬浮于等体积的pbs/5mmedta(室温)中。将30(30)ml的ficoll-paqueplus(fischerscientific,目录号11778538)吸移到leucosep管中,然后使30ml的猪pbmc血沉棕黄层/pbs/edta混合物在ficoll-paqueplus顶部分层。在室温下以800g离心25min后,从猪血浆和ficoll-paqueplus的界面处收集猪pbmc。将pbmc转移到新的离心管中,并补充冷的pbs/5mmedta。将细胞悬浮液在4℃下以350g离心10min,并弃去上清液。然后将细胞沉淀重新悬浮于0.2%的低渗盐水中,以溶解任何残余的红细胞。在30秒后,使细胞悬浮液等渗,并在4℃下以500g离心5min。然后将pbmc细胞沉淀重新悬浮于完全培养基(dmem和glutamax(thermofisher,目录号32430100),10%的马血清(30-2040tm),20μm的β-巯基乙醇(thermofisher目录号31350010[贮存液:50mm])中。试验也在完全培养基中进行。将pbmc以1×106个细胞/孔的细胞密度接种到24-孔板上。将本发明化合物溶于dmso中,并在完全培养基中进行一系列稀释。化合物实施例在试验中以在0.003μm至10μm范围内的8个不同的浓度使用,使得最终的dmso浓度为0.5%。为诱导细胞因子分泌,用0.01ng/ml至1ng/ml浓度范围内的lps(lps来自e.colik12;invivogen#tlrl-eklps)刺激细胞24小时。使用türk溶液(merckmillipore#1092770100)测定细胞生存力。使用基于兔抗猪tnfα抗体(rabbitanti-porcinetnfαantibody)的elisa读出装置测定暴露于lps的猪pbmc的细胞培养物上清液中所分泌的tnfα的量。elisa测定在384孔elisa板中进行,所述elisa板用在50mmph9.6的na2co3/nahco3缓冲液中的3μg/ml的兔抗猪tnfα抗体(biorad,ahp2397)以10μl/孔且在4℃下包被48h。在除去抗体并用50μl的清洗缓冲液(pbs,0.05%(v/v)tween20)将孔冲洗三次后,用50μl的封闭缓冲液(pbs,0.05%(v/v)tween20,1%(w/v)牛血清白蛋白)将孔在37℃下温育60min。其后,除去封闭缓冲液,并加入培养物上清液样品(20μl/孔)。在37℃下温育90min后,除去样品,并用50μl的清洗缓冲液将孔冲洗三次。将0.25μg/ml的在封闭缓冲液中的兔抗猪tnfα生物素缀合抗体(biorad,ahp2397b)加入到在37℃下温育60min的板(20μl/孔)中。在除去生物素化的抗体并用50μl的清洗缓冲液将孔冲洗三次后,在37℃下加入1:10.000稀释在封闭缓冲液中的extravidintm-碱性磷酸酶(sigma,e2636),20μl/孔,保持1h。在除去extravidintm-碱性磷酸酶并用50μl的清洗缓冲液将孔冲洗三次后,通过加入90μl/孔的显色缓冲液(于50mmph9.6的na2co3/nahco3,2mmmgcl2中的5mm的对硝基苯磷酸酯(pnpp))引发酶促反应/显色。记录在405nm波长下的光密度。对于动力学测量,数据点每隔5分钟记录一次,记录1小时,在2小时后进行终点测量。例如,10μm实施例化合物12抑制通过0.1ng/mllps刺激的牛pbmc的tnfα的生成。这由图10说明。室内尘螨诱导的犬过敏性皮炎的体内模型为评估本发明通式(i)的化合物的潜在抗过敏/抗炎症功效,使用室内尘螨致敏的比格犬(beagledogs)模型。其中,hmd致敏包括每隔大约2周,一系列地皮下注射hdm抗原(10μg,greerlaboratories,lenoir,nc,usa)和(0.2ml,invivogen,sandiego,ca921221,usa)作为佐剂。致敏过程通过皮内皮肤测试监测并确认。一旦狗对hdm皮内皮肤测试呈阳性,距离最后一次致敏一个月时,则局部施用hdm抗原(135μg),并刺入成年比格犬后腿内部的皮肤内(用2mm长的微型针),从而检测本发明化合物对过敏性皮炎症状如红斑和水肿的功效。有2组,每组4只动物:1组为安慰剂对照组,1组用实施例化合物12治疗。对照组用含有微纤维素的明胶胶囊口服治疗,而用实施例化合物12治疗的组用含有实施例化合物12和微纤维素的明胶胶囊口服治疗。在用hdm抗原攻击以前5天开始给予实施例化合物12或安慰剂,并持续至攻击以后2天。治疗频率为每天一次,在实施例化合物12的情况下,剂量为10mg/kg体重。在攻击开始以后30min以及在48h以内的不同的时间点,在两组中使用vas(视觉模拟评分法)评估红斑和水肿。分析血浆样品以确定与临床评估相关的所述化合物接触。在用实施例化合物12治疗后,水肿和红斑显著减轻。这由表4和表5,以及图11和图12说明。表4:用实施例化合物12与安慰剂治疗后的红斑(以vas为单位)表5:用实施例化合物12与安慰剂治疗后的水肿(以vas为单位)跳蚤过敏性皮炎的体内瘙痒模型为评估本发明通式(i)的化合物的潜在止痒功效,使用了跳蚤过敏性皮炎(fad)模型。在研究中仅纳入具有fad病史的成年狗。所述研究的生命期由两个阶段组成:瘙痒诱导阶段(2周)和随后的治疗阶段(2周)。在两个研究阶段期间,使狗每周两次感染栉首蚤属(ctenocephalides)跳蚤(第一次攻击用100只跳蚤/狗,所有的后续攻击用30只跳蚤/狗)。有2组,每组12只动物:1组为安慰剂对照组,1组用实施例化合物12治疗。对照组用含有微纤维素的明胶胶囊口服治疗,而用实施例化合物12治疗的组用含有实施例化合物12和微纤维素的明胶胶囊口服治疗。治疗频率为每天一次,在实施例化合物12的情况下,剂量为20mg/kg体重。从治疗后第一天开始,每到第三天,对狗进行一次记录,持续4小时,并且耗费在瘙痒行为上的时间测定为耗费在挠、舔、咬上的秒数。分析血浆样品以确定与临床评估相关的所述化合物接触。在用实施例化合物12治疗10天后,瘙痒显著减轻。这由表6和图13说明。表6:与安慰剂相比,在用实施例化合物12治疗后,瘙痒行为相对于基线的减轻(显示为在治疗后列出的天由基线的百分比变化)第4天第7天第10天第13天安慰剂对照组-12.0%-9.9%1.7%-20.0%实施例化合物12-26.7%5.7%-57.7%-48.0%图1:实施例化合物12对人单核细胞生成的dc中il-23的抑制。数据显示为具有标准偏差的平均值。图2:实施例化合物12对(a)咪喹莫特(r837)-或(b)cpg-a-刺激的人浆细胞样dc中inf-α的抑制。数据显示为具有标准偏差的平均值。图3:用实施例化合物11治疗lps诱导的炎症导致分泌的tnf-α量减少。数据显示为具有标准偏差的平均值。图4:用实施例化合物11(左)和12(右)治疗il-1β诱导的炎症导致分泌的tnf-α的量的剂量依赖性减少。数据显示为具有标准偏差的平均值。图5:实施例化合物11在类风湿性关节炎动物模型(佐剂诱导的大鼠模型)中的抗炎功效。基于疾病活动评分测定的类风湿性关节炎的显著和剂量依赖性抑制。数据对应于平均值+标准偏差。单因素anova方差分析,随后通过dunnett’s检验与cfa对照组多重比较分析;*p<0.05;**p<0.01;***p<0.001;****p<0.0001。图6:实施例化合物12在类风湿性关节炎动物模型(胶原抗体诱导的小鼠模型)中的抗炎功效。基于疾病活动评分测定的类风湿性关节炎的显著和剂量依赖性抑制。数据对应于平均值+标准偏差。通过单因素anova方差分析,随后多重比较分析(dunnett’s检验)来计算胶原抗体(ak)对照组和治疗组之间的统计显著性(*p<0.05;**p<0.01;***p<0.001;****p<0.0001)。图7:实施例化合物12对lps诱导的通过犬pbmc的tnfα生成的抑制。数据显示的是具有标准偏差的平均值。图8:实施例化合物12对通过由1μg/mllps诱导的牛pbmc的tnfα生成的剂量依赖性抑制(动力学测定)。数据显示的是三份生物材料的具有标准偏差的平均值,每份测量两次。由该曲线测定的ic50值为120nm。图9:实施例化合物12对通过由1μg/mllps诱导的牛pbmc的tnfα生成的剂量依赖性抑制(动力学测定)。数据显示的是三份生物材料的具有标准偏差的平均值,每份测量两次。由该曲线测定的ic50值为70.5nm。图10:10μm的实施例化合物12对通过由0.1ng/mllps诱导的猪pbmc的tnfα生成的抑制(动力学测定)。数据显示的是三份生物材料的具有标准偏差的平均值,每份测量两次。图11:用实施例化合物12治疗室内尘螨诱导的犬过敏性皮炎模型导致红斑(a)减轻。数据显示为具有标准偏差的平均值。图12:用实施例化合物12治疗室内尘螨诱导的犬过敏性皮炎模型导致水肿(b)减轻。数据显示为具有标准偏差的平均值。图13:实施例化合物12在跳蚤过敏性皮炎的动物模型中的止痒功效。数据表示为与中值对应的由基线的百分比变化。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1