制备光学活性贝前列素的方法与流程
市售的贝前列素钠用于治疗外周动脉疾病(drugs,2002,62,107-133)且自2007年起其也用于治疗肺动脉高压(pah)(j.am.coll.cardiol.,2004,43,56s-61s)。
考虑到其化学结构,贝前列素钠是一种外消旋化合物即四种异构体的混合物。
*chinoin名称,**toray名称(也在cas中公开)
toray公司的贝前列素合成的原料为环戊二烯(2)。环戊二烯被溴化,二溴环戊烯(3)与三溴苯酚反应。自所得化合物形成的grignard试剂在cui催化剂存在下转化成外消旋三环(rac-5)*,通过prins反应自其制备外消旋二羟基衍生物(rac-6)。两个羟基通过缩醛进行保护(rac-7),上链通过甲酰基丙酸甲酯(8)构建并氢化,将缩醛保护基裂解,对两个羟基进行选择性保护,将伯羟基释放并氧化成醛(rac-9)。下链通过使用外消旋膦酸酯(10)的horner-wadsworth-emmon(hwe)反应构建,所得外消旋烯酮衍生物(rac-11)的酮基被还原成羟基,分离异构体(rac-12),酯基被水解并分离酸的钠盐(tetrahedron,1999,55,2449-2474)(方案1)。
*结构显示相对立体化学
方案1
对于在进一步治疗领域中的应用,最有效的形式即活性贝前列素(314-dbps=1α2/1)可按光学纯形式得到是有必要的。
根据toray公司公开的方法,通过使自外消旋三环(rac-5)所得grignard试剂与二氧化碳反应并通过所得粗外消旋酸用手性胺((+)-顺式-n-苄基-2-(羟基甲基)环己胺)拆分来制备光学活性贝前列素钠(1α2/1)以得到光学活性酸(14)(tetrahedronasymmetry,1999,10,4099-4105)。
在光学活性酸(14)的双键上,通过prins反应形成羟基和羟基甲基,通过催化氢化除去溴代基团,使酸官能团酯化(15),游离羟基用thp基团保护,将酯基还原为醇,然后氧化成醛(16),上链通过wittig反应构建,将酸转化成甲酯,然后除去thp基团(17),使上链中的双键饱和,羟基被选择性保护,释放伯羟基,然后氧化成醛,得到rac-9的光学活性形式(9)。使醛与手性膦酸酯(s-10)反应,其通过已经描述的化学步骤转化成光学活性贝前列素钠异构体。制备全部四种贝前列素钠异构体,但是该公开既没有披露盐的制备,也没有披露盐的特征(heterocycles,2000,53,1085-1110)(方案2)。
方案2
对于手性膦酸酯(s-10)的制备,外消旋2-甲基-4-己炔酸(rac-18)用手性胺(奎宁、辛可尼定、1-苄基氨基-2-羟基乙基-环己烷(顺式-胺))拆分(18)(tetrahedronasymmetry,2000,11,2981-2989)(方案3)。
方案3
专利申请wo2012/174407a1公开了光学活性贝前列素的制备。合成的原料为光学活性corey-内酯(19),其在几个步骤中转化成经保护的二氢呋喃衍生物(20)。20在脱羧和芳构化后与3-甲氧羰基-2-吡喃酮(21)进行由
lantanida催化(eu(hfc)3)的diels-alder反应,得到含有苯并呋喃环的中间体(22)(方案4)。
方案4
为了构建上链,将22的甲酯基分两步转化成醛,自醛(23)与(1-重氮基-2-氧代丙基)-膦酸二甲酯(24)得到乙炔衍生物(25),使其与重氮乙酸乙酯(26)反应,最后通过催化氢化(27)使叁键饱和(方案5)。
方案5
释放经保护的二醇27的伯羟基并氧化成醛,使醛28与s-10膦酸酯反应,用硼氢化钠氯化铈(iii)试剂还原烯酮29的氧代基团或在(r)-(+)-cbs催化剂存在下用儿茶酚硼烷或硼烷-二甲基硫醚试剂选择性还原,得到(30),自仲醇除去保护基,酯用氢氧化钠的甲醇溶液转化成钠盐(方案6)。
方案6
钠盐的物理特性尚未公开。
自活性贝前列素酸还在乙酸乙酯中用氢氧化钾的乙醇溶液制备了钾盐。将钾盐从乙醇水溶液中重结晶。
专利申请wo2013/040068a1公开的方法的原料是光学活性环戊烯酮衍生物31,其用溴苯酚衍生物32烷基化。使芳基醚33环化,对氧代基团进行还原,得到醇(34)。仲羟基用乙酰基保护且对双键进行由pdcl2催化的氧化转化,得到衍生物35。对双键进行臭氧解,然后与三苯基膦反应,由此得到醛9(方案7)。
方案7
使醛9与光学活性r-10膦酸酯反应,烯酮r-11的氧代基团在(r)-cbs催化剂存在下用硼烷-四氢呋喃试剂选择性还原,然后将酯基水解,得到贝前列素酸的α1/1异构体,其不是活性异构体。
该专利要求保护经由上述化学步骤通过使用相应的膦酸酯对映异构体(s-10和r-10)来制备各种贝前列素酸异构体。
贝前列素钠盐的制备在说明书中没有描述。
根据专利说明书wo2015/179427的方法的原料为手性卤代衍生物35,其在几个步骤中转化成经保护的二醇36。将二醇36的游离羟基氧化,得到醛,然后使醛与手性膦酸酯s-10反应。
r1=甲基-或乙基-
r2=h或适于保护醇的基团
在r2=h且r2=叔丁基-二甲基甲硅烷基的情况下还进行了立体选择性还原。将活性酯37水解成酸。将酸(固体泡沫状物)转化成结晶性钾盐。
本申请主题为制备式i光学活性贝前列素及其盐的方法:
其特征在于
使通式ii外消旋贝前列素酯水解:
其中r表示c1-4直链或支链烷基,
使所得式iii外消旋贝前列素酸结晶:
对所得式iiia和iiib贝前列素酸对映异构体进行酯化:
使所得式iva和ivb贝前列素酯对映异构体与手性酸或酸衍生物反应:
其中r’表示c1-4直链或支链烷基,
分离所得二酰基-贝前列素酯非对映异构体via和vib:
其中r’具有与上述相同的含义且r2意指含有手性碳原子的酸残基,
使式via贝前列素酯水解且将所得式i光学活性贝前列素酸以结晶形式分离且若需要,则转化成其盐。
根据本申请优选实施方案,式ii化合物的水解在水可混溶性有机溶剂中用无机碱的水溶液进行,可使用醇(甲醇、乙醇、异丙醇)或水可混溶性醚(乙醚、四氢呋喃、二噁烷)或其它水可混溶性溶剂(例如乙腈)作为溶剂且可使用氢氧化钾、氢氧化钠作为无机碱。
结晶在极性-非极性溶剂混合物中且优选在乙酸乙酯:己烷混合物中进行且反复结晶若干次。
可使用手性酸或酸衍生物的光学纯对映异构体作为手性试剂,例如苹果酸、氨基酸、酒石酸或酒石酸衍生物(例如二苯甲酰基酒石酸)、樟脑酸或樟脑酸衍生物(例如樟脑磺酸)、薄荷氧基乙酸、α-甲氧基苯基乙酸、α-甲氧基-α-三氟甲基苯基乙酸、2-苯基丙酸、扁桃酸或扁桃酸衍生物(例如氯扁桃酸、乙酰基-氯扁桃酸且优选r-乙酰基-氯扁桃酸)。
根据本申请通过色谱法且优选通过常压硅胶色谱法分离通式via和vib非对映异构体。
可使用多组分梯度混合物作为洗脱剂进行色谱。可使用饱和烃(戊烷、己烷、庚烷、异辛烷、环己烷、甲基环己烷)或芳族烃(甲苯)或卤代烃(二氯甲烷)作为非极性溶剂组分。可使用醇(甲醇、乙醇、异丙醇)、酯(乙酸甲酯、乙酸乙酯、乙酸异丙酯)、醚(乙醚、甲基叔丁基醚)或酮类(丙酮、甲基乙基酮、甲基异丁基酮)溶剂混合物作为极性组分。优选使用二氯甲烷:乙酸乙酯混合物作为洗脱剂。
根据本申请优选实施方案,式i光学活性贝前列素酸以结晶形式分离。使用丙酮:水和二氯甲烷:二异丙基醚:己烷溶剂进行结晶。若需要,则将式i贝前列素酸转化成其盐。
本申请优选实施方案详述如下:
该方法的原料为外消旋贝前列素酯(rac-bp-酯,ii),其可根据专利hu-227158b1或专利申请wo2003/011849制备。
rac-贝前列素酯(ii)
外消旋贝前列素酯为4种异构体的约1:1比例混合物。
异构体α2/1-α2/2和α1/1-α1/2为对映异构体,而异构体α2/1-α1/1和α2/2-α1/2为非对映异构体。
本申请方法的基础是具有不同物理特性的非对映异构体可通过物理方法(例如结晶、色谱法)分离。
然而,对映异构体的物理特性是相同的,唯一的区别是它们使线性偏振光的平面在相反的方向上旋转。
通过简单的物理方法分离对映异构体是不可能的,它们的分离需要手性辅助材料。
在本申请方法中首先分离非对映异构体对。
出于该目的,将含有4种异构体的外消旋贝前列素酯(ii)水解成含有4种异构体的外消旋贝前列素酸(ii)。
贝前列素酸的非对映异构体通过反复结晶分离。自己烷:乙酸乙酯溶剂混合物进行结晶。
反复结晶后,外消旋贝前列素酸仅含有α2/1贝前列素酸及其α2/2对映异构体。
rac-贝前列素酸(α2/1和α2/2异构体)
由于对映异构体的分离需要手性辅助材料,因此首先用碘甲烷对含有α2/1和α2/2对映异构体对的rac-贝前列素酸进行酯化,然后rac-贝前列素酯(α2/1和α2/2异构体)的游离羟基用手性酸即r-乙酰基-氯扁桃酸(v)酯化。
用手性酸进行酯化的二酰基-贝前列素酯非对映异构体已经可通过物理方法分离且在此通过色谱法分离。
使用二氯甲烷:乙酸乙酯溶剂混合物进行色谱分离。
色谱的主要级分是自活性贝前列素酯(iva)与r-乙酰基-氯扁桃酸形成的二酯(二酰基-贝前列素酯(α2/1))。
二酰基-贝前列素酯(α2/1)
对酯基进行水解,得到活性贝前列素酸(贝前列素酸(α2/1),i)。
通过反复结晶可将结晶性活性贝前列素酸的异构体杂质的量降低至满足由质量要求确定的限度的值。
自丙酮-水和二氯甲烷:二异丙基醚:己烷混合物进行结晶。
若需要,则可将活性结晶性贝前列素酸转化成其盐。
该方法在方案8中显示。
方案8
本申请通过以下实施例说明,而非使本申请所要求保护的方案限于实施例所描述的方案。
实施例
自外消旋贝前列素酯(rac-bp-酯)制备活性贝前列素钠(1α2/1)
a.)rac-贝前列素酸
(1r*,2r*,3as*,8bs*)-2,3,3a,8b-四氢-2-羟基-1-[(e,3s*,4rs)-3-羟基-4-甲基-1-辛烯-6-炔基]-1h-环戊并[b]苯并呋喃-5-丁酸
将400.0grac-bp-酯溶于1.5l四氢呋喃中,在室温在惰性气氛中在搅拌下向溶液中添加6l0.5m氢氧化钠溶液。当水解结束时,反应混合物用水稀释并用甲基叔丁基醚洗涤。水溶液用1m硫酸氢钠溶液酸化至ph≤3。酸性水溶液用甲基叔丁基醚萃取。合并的有机相用饱和盐溶液洗涤至中性,用硫酸钠干燥并蒸发。将经蒸发的浓缩物溶于乙酸乙酯中并用己烷结晶。
产率:290.0g(75%)。
b.)rac-贝前列素酸(α2/1和α2/2异构体)(rac-bp-酸(α2/1和α2/2异构体))
(1r*,2r*,3as*,8bs*)-2,3,3a,8b-四氢-2-羟基-1-[(e,3s*,4s*)-3-羟基-4-甲基-1-辛烯-6-炔基]-1h-环戊并[b]苯并呋喃-5-丁酸
在50℃将290grac-bp-酸溶于2.9l乙酸乙酯中,将溶液冷却至室温且在搅拌下向其中添加2.9l己烷。大量晶体析出后,向悬浮液中再添加1.45l己烷,然后将其冷却至0-10℃并在该温度搅拌15分钟。滤出晶体并洗涤。
使滤器湿晶体再次结晶。
反复结晶直到通过hplc确定rac-贝前列素酸(α1/1和α1/2异构体)的量降低至≤0.5%。
为了满足所需质量要求而需要进行10次结晶。
产率:82.0g(28.3%)。
c.)rac-贝前列素酯(α2/1和α2/2异构体)(rac-bp-酯(α2/1和α2/2异构体))
(1r*,2r*,3as*,8bs*)-2,3,3a,8b-四氢-2-羟基-1-[(e,3s*,4s*)-3-羟基-4-甲基-1-辛烯-6-炔基]-1h-环戊并[b]苯并呋喃-5-丁酸甲酯
将82grac-bp-酸(2/1和2/2异构体)溶于410ml二甲基甲酰胺中,向其中添加49.8g碳酸钾和25.6ml碘甲烷。将混合物在40℃搅拌直到达到所期望的转化。当反应完成时,将混合物倾倒在酸性水上并用甲苯萃取产物。合并的有机相用1m碳酸氢钠溶液、水和饱和盐溶液洗涤,用硫酸钠干燥并蒸发。
产率:84.0g(99.0%)。
d.)二酰基-贝前列素酯(α2/1异构体)(二酰基-bp-酯(α2/1))
(1r,2r,3as,8bs)-2,3,3a,8b-四氢-2-[2r-乙酰氧基-2-(2-氯苯基)-乙酰氧基]-1-[(e,3s,4s)-3-[2r-乙酰氧基-2-(2-氯苯基)-乙酰氧基]-4-甲基-1-辛烯-6-炔基]-1h-环戊并[b]苯并呋喃-5-丁酸甲酯
在惰性气氛中将84.0grac-bp-酯(α2/1和α2/2异构体)溶于1.68l二氯甲烷(dcm)中,向溶液中添加9.95g二甲基氨基吡啶(dmap)和107.1gr-乙酰基氯扁桃酸且搅拌混合物直到溶解。完全溶解后,将反应混合物冷却至(-)-10℃且添加100.8g二环己基碳二亚胺(dcc)。在不冷却的情况下搅拌反应混合物直到达到所期望的转化。当反应结束时,过量的二环己基碳二亚胺用1m盐酸破坏,滤出所析出的物质且用乙酸乙酯洗涤。合并的有机相先后用1m碳酸氢钠溶液和饱和盐溶液洗涤,用硫酸钠干燥并蒸发。经蒸发的浓缩物在硅胶柱上使用二氯甲烷:乙酸乙酯=20:1和二氯甲烷:乙酸乙酯=5:1洗脱剂混合物进行色谱分离。二酰基-bp-酯(α2/1)异构体在二酰基-bp-酯(α2/2)异构体后洗脱。蒸发含有二酰基-bp-酯(α2/1)异构体的主要级分。
产率:83.0g(48.9%)。
r-乙酰基-氯扁桃酸的制备
2r-乙酰氧基-2-(2-氯苯基)乙酸
在室温在搅拌下将100gr-氯扁桃酸溶于95ml乙酸酐中。完全溶解后,将反应混合物真空浓缩,向浓缩物中添加甲苯,然后真空蒸馏出甲苯。
在室温将经蒸发的浓缩物溶于二异丙基醚和己烷的混合物中。搅拌溶液直到结晶开始,然后添加额外量的己烷。将悬浮液冷却至0℃以完成结晶。
产率:112.0g(91.4%)。
e.)贝前列素酸(α2/1异构体)(bp-酸(α2/1))
(1r,2r,3as,8bs)-2,3,3a,8b-四氢-2-羟基-1-[(e,3s,4s)-3-羟基-4-甲基-1-辛烯-6-炔基]-1h-环戊并[b]苯并呋喃-5-丁酸
将83.0g二酰基-bp-酯(α2/1)溶于1l甲醇中。向溶液中添加0.84l1m氢氧化钠溶液且搅拌混合物直到水解进行。当反应结束时,真空除去甲醇。经浓缩的溶液用1m硫酸氢钠溶液酸化并用乙酸乙酯萃取。合并的有机相用饱和盐溶液洗涤,用硫酸钠干燥并蒸发。
将经蒸发的浓缩物溶于256ml丙酮中并在室温用2.57l水结晶。滤出晶体且将滤器湿产物在35-40℃重复溶于143ml丙酮中且在冷却至室温后用1.43l水结晶。滤出晶体并干燥。
在40℃将干燥的晶体悬浮于70ml二氯甲烷和525ml二异丙基醚的混合物中,搅拌10分钟并缓慢冷却至25℃。滤出不溶解的晶体。
在室温向滤液溶液中滴加约1l己烷,然后将晶体悬浮液冷却至0℃以完成结晶。滤出晶体,洗涤并干燥。
产率:30.0g(75.6%),熔点:61-64℃
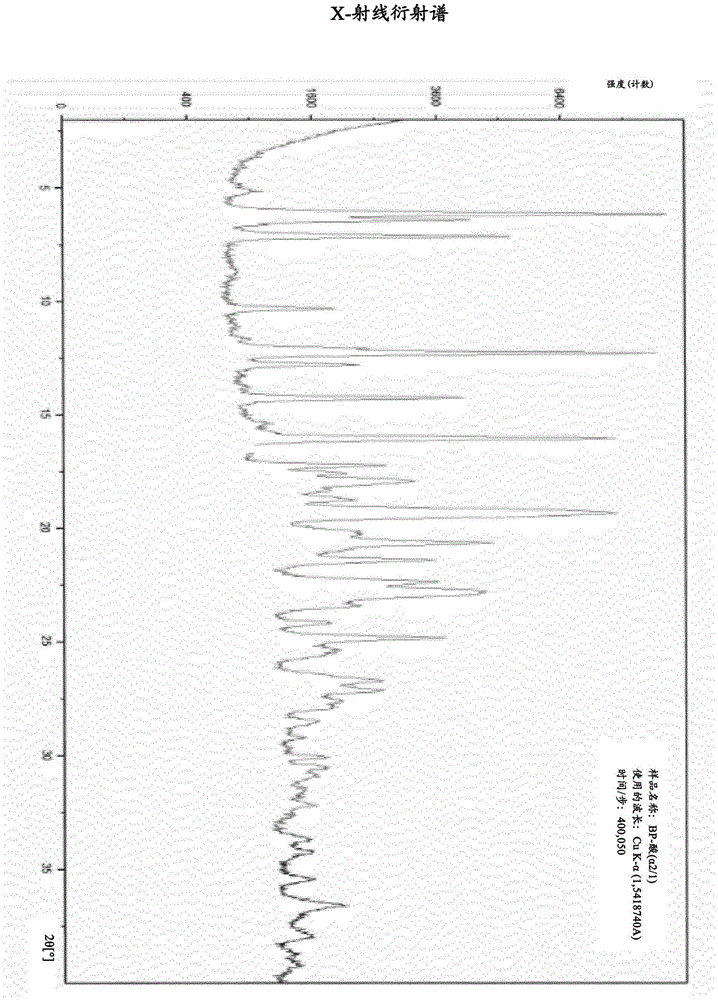
结晶性光学活性贝前列素酸的x射线衍射谱(在图1中显示)的特征峰:
x射线衍射谱的测量条件:
起始位置[°2θ]:2.0084
结束位置[°2θ]:39.9864
测量温度[℃]:25.00
阳极材料:cu
上述物质的dcs曲线在图1中显示。
dsc测量条件:
仪器:
mettlertoledodsc1staresystem
starebasicv9.30
方法:
起始温度:150℃
结束温度:250℃
加热速度:10℃/min、5℃/min、2℃/min
重量:2-6mg
多孔氧化铝坩埚(40μl)
- 还没有人留言评论。精彩留言会获得点赞!