利用负载型混杂茂金属催化剂的高密度乙烯类聚合物及其制备方法与流程
本发明涉及一种利用负载型混杂茂金属催化剂的高密度乙烯类共聚物及其制备方法,更详细而言,涉及一种高密度乙烯类聚合物,其具有高分子量,并且共聚单体(comonomer)的含量相对集中在高分子量体中,因而表现出优异的机械特性和耐化学性,并且具有宽分子量分布和长链支化结构,因此具有高熔融流动性。
背景技术:
聚乙烯树脂由于分子量以及密度而机械、热学特性受到影响,因此,应用领域也不同。一般,聚乙烯聚合物的密度越低,透明性和冲击强度等越好,但是耐热性、硬度以及弯曲弹性率等物理性质变差,并且耐化学性也变差。
反之,聚乙烯聚合物的密度越高,耐热性、硬度以及弯曲弹性率等物理性质就越好,并且耐化学性也提升,但是透明性以及冲击强度变差。因此,当制造利用乙烯共聚物的注塑产品,特别是食品容器等时,相当难以制造冲击强度优异的同时耐化学性也优异的注塑产品。特别是市场上所需的食品容器等注塑产品要求高耐冲击性,因此对这种技术的需求非常大。
通过各种成型方法,高密度聚乙烯聚合物以多种用途被提供。例如,作为薄膜成型体的代表性的方法,有吹胀法(inflationmethod),该法熔融高密度聚乙烯聚合物,吹入空气,并从模具挤出,从而吹胀出熔融聚合物挤出物。并且,作为获得想要的形状的成型体的方法,有吹塑成型法(blowmoldingmethod),该法将熔融的高密度聚乙烯聚合物吹入模具的空洞中后,将空气吹入模具空洞中的熔融树脂中,从而使熔融树脂膨胀并挤压空洞内壁,由此在空洞内形成熔融聚合物。此外,有注射成型法,该法将熔融的高密度聚乙烯聚合物压入空洞内,从而填充空洞。
如此,高密度聚乙烯聚合物有多种成型方法,只是这些方法的共同之处在于,首先对聚乙烯聚合物进行加热,从而形成熔融状态,并使其成型。因此,在高密度聚乙烯聚合物的成型加工中,高密度聚乙烯聚合物的加热、熔融时的行为,即熔融特性是极为重要的物理性质。
特别是,在挤出、压缩、注射或者旋转成型等成型中,熔融特性,尤其是高密度聚乙烯类聚合物的熔融流动性是决定令人满意的成型加工性的根本性的物理性质。本发明中所称的成型加工性并不局限于挤出、压缩、注射或者旋转成型时的加工性。
一般,可以认为,mi、mfi、mfr越大,熔融流动性就越优异。然而,现实中各个成型方法中所要求的成型材料聚合物的性状都有不同,因此,作为表示成型加工性的标准而使用的指标,根据各个成型方法而不同。例如,在注射成型法中,为了获得具有耐冲击性的成型品,倾向于使用分子量分布窄的高密度聚乙烯聚合物。
以往,用于挤出、压缩、注射或者旋转成型等的高密度聚乙烯聚合物一般使用钛类的齐格勒-纳塔催化剂或者铬类催化剂来制备。
然而,使用这类催化剂制备的高密度聚乙烯聚合物的分子量分布宽,因此能够提高流动性,但是由于混入有低分子量成分,耐冲击性等机械物理性质明显降低,并且共聚单体(comonomer)集中分布于低分子量体中,因此耐化学性降低。因此无法在保持良好的机械物理性质的同时实现注射成型中的高速化。
为了解决这种问题,对茂金属催化剂进行了大量的研究。美国专利第6525150号公开了一种茂金属催化剂,其利用茂金属的均匀的活性点,能够制备分子量分布窄,并且,对于共聚物来说,能够制备共聚单体分布均匀的树脂。然而,由于其分子量分布窄,虽然机械强度优异,但是成型加工性差。
如前所述,通常对于单一的茂金属催化剂来说,由于均匀的活性点,分子量分布窄,因此成型加工性并不好,所以在重视机械物理性质与成型性的均衡的高密度聚乙烯聚合物领域,茂金属催化剂未能得到充分的应用开发。
为了解决这种问题,提出较多的是通过使用多个反应器,或者混合多样种类的茂金属催化剂的方法,使分子量分布变宽。
然而,当采用这种使分子量分布变宽的方法时,虽然成型性得以提升,但是不可避免地降低其它物理性质,因而无法获得通过使分子量分布变窄而得到的机械强度等物理性质优异的高密度聚乙烯聚合物。
此外,公开了通过保持催化剂的固有粘度来提高熔融张力的方法,但其未能改善熔融流动性的降低,因此存在难以高速成型的问题。
为了解决上述的茂金属催化剂的问题,利用将lcb(longchainbranch,长支链)作为侧链引入到聚合物主链的催化剂,改善了聚合物的熔融流动性,但是耐冲击性等机械物理性质明显低于使用通常的茂金属催化剂的情况。
此外,作为另一种方案,公开了利用对于共聚单体的反应性不同的催化剂来制备具有双峰(bimodal)分子量分布的聚烯烃的方法。然而通过这种方式制备的具有双峰分子量分布的聚烯烃虽然熔融流动性得以提升,但是由于具有不同的分子量分布,降低了相容性。因此难以获得加工后具有均匀物理性质的产品,并且机械强度降低。
为了改善利用茂金属催化剂来制备的高密度聚乙烯聚合物的机械特性和熔融流动性,公开了很多方法,但是大部分仅公开了对于线性低密度聚烯烃的解决方法。此外,茂金属具有随着共聚单体的浓度降低,活性也倾向于降低的特性,因此在制备高密度聚乙烯时活性低,并不经济。
即便是在制备低密度聚烯烃时具有优异的活性和加工性特征的催化剂,在制备高密度聚烯烃时也因活性低而不经济,特别是在气相工序中形成大量的微细粒子,因而难以进行稳定的操作。
在气相反应器中,活性是重要的因子,然而因活性低而生成大量的微细粒子,并由此产生大量的静电而粘附在反应器的壁面上,妨碍热传递,从而降低聚合温度,并且粘附在壁面上的微细粒子继续变大,所以在以往会使生产中断。
一直需要用来制备解决上述问题并且机械强度和熔融流动性优异且活性高的高密度聚烯烃聚合物的催化剂,也需要对此进行改善。
技术实现要素:
技术问题
本发明的目的在于解决上述的所有问题。
本发明旨在提供一种高密度乙烯类聚合物及其制备方法,所述高密度乙烯类聚合物同时满足以往的高密度乙烯类聚合物未能实现的机械特性与耐化学性、优异的成型加工性。
本发明的另一目的在于,提供一种高密度乙烯类聚合物及其制备方法,所述高密度乙烯类聚合物在后述的负载型混杂茂金属催化剂存在的情况下制备,高分子量体中的共聚单体(comonomer)的含量高,低分子量体中的共聚单体的含量低,因此冲击强度、弯曲强度、耐应力开裂性(escr)、熔融张力(melttension)优异,并且具有单峰分子量分布。
本发明的另一目的在于,提供一种高密度乙烯类聚合物及其制备方法,所述高密度乙烯类聚合物具有宽分子量分布和长链支化结构,因此在进行挤出、压缩、注射、旋转成型等加工时负荷小,具有优异的生产性。
技术方案
用于达成上述的本发明的目的,并且实现后述的本发明的特征效果的本发明的特征构成如下所述。
根据本发明的一实施方式,提供一种高密度乙烯类聚合物,其密度为0.940至0.970g/cm3,mi为0.1至50g/10min,mfr为37至100。
根据本发明的一实施方式,提供一种高密度乙烯类聚合物,其按照astmd1893测定的耐环境应力开裂(escr)中出现5个开裂的时间如下:当密度为0.950至0.960g/cm3且mi为1.0至3g/10min时,为16小时以上;当密度为0.950至0.965g/cm3且mi为6.0至7.0g/10min时,为10小时以上;当密度为0.955至0.960g/cm3且mi为8.0至9.0g/10min时,为9小时以上。
本发明涉及的聚烯烃聚合用负载型混杂茂金属催化剂提供一种用于制备聚烯烃的催化剂组合物,所述催化剂组合物包括由下述化学式1表示的至少一种第一茂金属化合物、由下述化学式2表示的至少一种第二茂金属化合物以及至少一种助催化剂化合物。
化学式1:
在所述化学式1中,m1是元素周期表中的第4族过渡金属,x1、x2各自独立地为卤素原子中的任意一种,r1至r12各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r1至r12能够彼此连接以形成环,与r1至r5结合的环戊二烯以及与r6至r12结合的茚是具有不同结构的不对称结构,所述环戊二烯与所述茚未连接,因此形成非交联结构。
化学式2:
在所述化学式2中,m2是元素周期表中的第4族过渡金属,x3、x4各自独立地为卤素原子中的任意一种,r13至r18各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r13至r18能够彼此连接以形成环,r21至r26各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r21至r26能够彼此连接以形成环,r19、r20各自独立地为被取代或未被取代的碳原子数1至20的烷基,r19、r20能够彼此连接以形成环,与r13至r18结合的茚以及与r21至r26结合的茚具有彼此相同的结构或者不同的结构,所述与r13至r18结合的茚以及所述与r21至r26结合的茚彼此与si连接,因此形成交联结构。
有益效果
本发明提供的负载型混杂茂金属催化剂的特性在于,制备高密度乙烯类聚合物时的活性高。此外,在所述催化剂存在的情况下制备的高密度乙烯类聚合物的特性在于,熔融流动性优异,同时冲击强度、弯曲强度、耐环境应力开裂性、熔融张力也优异。
附图说明
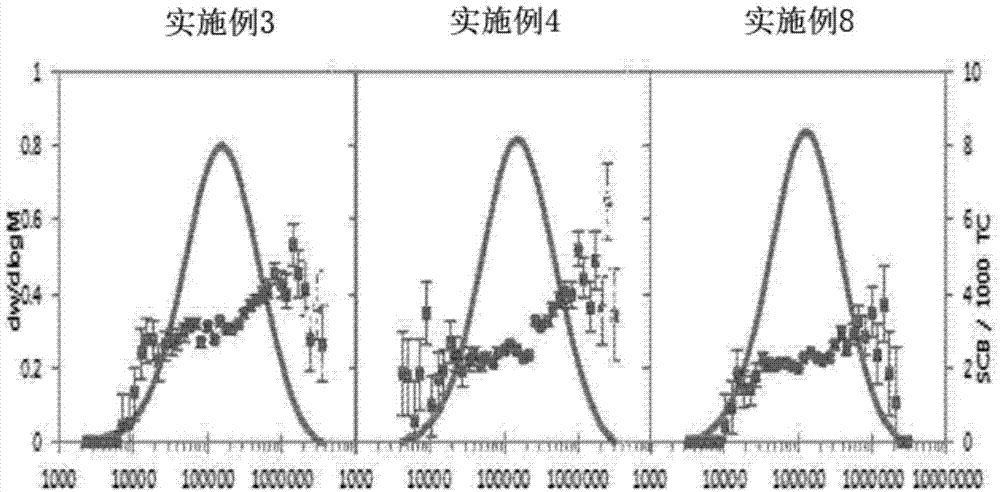
图1是为确认实施例3、4、8中所制备的共聚物的共聚单体分布而分析gpc-ir的曲线图。
图2是为确认比较例5、8中所制备的共聚物的共聚单体分布而分析gpc-ir的曲线图。
图3对实施例4中制备的共聚物与比较例9至11中制备的共聚物,比较了基于压力的注射成型性。
图4对实施例1、2、3、4、6、7、8中制备的共聚物与比较例9至17中制备的共聚物,比较了基于mi的注射成型性。
图5对实施例1、2、3、4、6、7、8中制备的共聚物与比较例9至17中制备的共聚物,比较了基于mi的冲击强度。
图6比较了实施例1、4中制备的共聚物与比较例9至11中制备的共聚物的加工性。
图7比较了实施例4中制备的共聚物与比较例9至11中制备的共聚物的流变性。
具体实施方式
后述的对于本发明的详细说明,参照了例示出能够实施本发明的特定实施例的附图。详细地说明这些实施例,以便本领域技术人员能够充分实施本发明。应理解为,本发明的各种实施例虽彼此不同,但无需相互排斥。例如,这里所记载的特定形状、结构以及特性与一个实施例相关,在不超出本发明的精神以及范围的情况下,可以由其他实施例实现。另外,应理解为,各公开的实施例内的个别构成要素的位置或配置在不超出本发明的精神以及范围的情况下可以变更。因此,后述的详细说明并非旨在限定本发明,准确地说,本发明的范围仅以权利要求书和其等同的所有范围限定。附图中的相似的附图标记在多个方面表示相同或相似的功能。
下面,参照附图详细地说明本发明的优选实施例,以便本领域技术人员能够容易地实施本发明。
本发明包括在负载型混杂茂金属催化剂存在的情况下聚合的高密度乙烯类聚合物。
本发明中利用的负载型混杂茂金属催化剂分别独立地包含一种以上的第一及第二茂金属化合物以及一种以上的助催化剂化合物。
本发明涉及的过渡金属化合物即第一茂金属化合物可以如下述化学式1所示。
第一茂金属化合物的作用在于,在混杂负载型催化剂中表现出高活性,并且提升所制备的聚合物的熔融流动性。
所述第一茂金属化合物具有共聚单体(comonomer)的混入度低,并且形成低分子量体的特征,因此提升加工聚合物时的加工性。
此外,由于共聚单体的混入少,形成高密度,在制备成高密度时也表现出高活性。
所述第一茂金属化合物为具有不同配体的不对称结构和非交联(unbridged)结构,因此形成共聚单体因难以接近催化剂活性点的立体障碍,从而起到减少共聚单体的混入的作用,并且在制造负载型混杂茂金属时使其表现出加工性和高催化剂活性。
化学式1
在所述化学式1中,m1是元素周期表中的第4族过渡金属,x1、x2各自独立地为卤素原子中的任意一种,r1至r12各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r1至r12可以彼此连接以形成环,与r1至r5结合的环戊二烯以及与r6至r12结合的茚是具有不同结构的不对称结构,所述环戊二烯与所述茚未连接,因此可以形成非交联结构。
将诸如本发明中所述化学式1中的与r1至r5结合的环戊二烯、与r6至r12结合的茚以及下述化学式2中的与r13至r18结合的茚、与r21至r26结合的茚的与过渡金属(化学式1及2中的m1及m2)进行配位键结合的离子或分子称为配体(ligand)。
在本发明中,除非有特别的提及,否则所述“取代”表示氢原子被卤素原子、碳原子数1至20的烃基,碳原子数1至20的烷氧基、碳原子数6至20的芳氧基等取代基取代。此外,除非有特别的提及,否则所述“烃基”表示直链型、支链型或者环型的饱和或者不饱和烃基,所述烷基、烯基、炔基等可以是直链型、支链型或者环型。
在具体例中,由所述化学式1表示的过渡金属的化合物的示例可以是下述结构的过渡金属化合物、它们的混合物等,但是并不仅限于此。
化学式1-1:
化学式1-2:
化学式1-3:
化学式1-4:
化学式1-5:
化学式1-6:
化学式1-7:
化学式1-8:
化学式1-9:
化学式1-10:
化学式1-11:
化学式1-12:
化学式1-13:
化学式1-14:
化学式1-15:
化学式1-16:
化学式1-17:
化学式1-18:
化学式1-19:
在所述过渡元素化合物中,m是元素周期表中的第4族过渡金属,例如铪(hf)、锆(zr)、钛(ti)等,me是甲基。
本发明涉及的过渡金属化合物即第二茂金属化合物可以如下述化学式2所示。
第二茂金属化合物的作用在于,在混杂负载型催化剂中表现出较高的共聚单体混入度,提升所制备的聚合物的机械物理性质。
所述第二茂金属化合物具有共聚单体的混入度高,形成高分子量体,使共聚单体集中分布在高分子量体中的特征,因此提升冲击强度、弯曲强度、耐环境应力开裂性、熔融张力。此外,第二茂金属化合物形成长链支化结构,从而提升高分子量的高密度乙烯类聚合物的熔融流动性。
所述第二茂金属化合物是具有多种配体的对称结构或者不对称结构以及交联结构,因此形成共聚单体易于接近催化剂活性点的立体障碍,从而起到增加共聚单体的混入的作用。
化学式2:
在所述化学式2中,m2是元素周期表中的第4族过渡金属,x3、x4各自独立地为卤素原子中的任意一种,r13至r18各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r13至r18可以彼此连接以形成环,r21至r26各自独立地为氢原子、被取代或未被取代的碳原子数1至10的烷基、被取代或未被取代的碳原子数6至20的芳基、被取代或未被取代的碳原子数7至40的烷基芳基,r21至r26可以彼此连接以形成环,r19、r20各自独立地为被取代或未被取代的碳原子数1至20的烷基,r19、r20可以彼此连接以形成环,与r13至r18结合的茚以及与r21至r26结合的茚可以是相同的结构或者不同的结构,与r13至r18结合的茚以及与r21至r26结合的茚彼此与si连接,因此可以形成交联结构。
在本发明中,除非有特别的提及,否则所述“取代”表示氢原子被卤素原子、碳原子数1至20的烃基,碳原子数1至20的烷氧基、碳原子数6至20的芳氧基等取代基取代。此外,除非有特别的提及,否则所述“烃基”表示直链型、支链型或者环型的饱和或者不饱和烃基,所述烷基、链烯基、炔基等可以是直链型、支链型或者环型。
在具体例中,由所述化学式2表示的过渡金属的化合物的示例可以是下述结构的过渡金属化合物、它们的混合物等,但是并不仅限于此。
化学式2-1:
化学式2-2:
化学式2-3:
化学式2-4:
化学式2-5:
化学式2-6:
化学式2-7:
化学式2-8:
化学式2-9:
化学式2-10:
化学式2-11:
化学式2-12:
化学式2-13:
化学式2-14:
化学式2-15:
化学式2-16:
化学式2-17:
化学式2-18:
化学式2-19:
化学式2-20:
化学式2-21:
化学式2-22:
化学式2-23:
化学式2-24:
化学式2-25:
化学式2-26:
化学式2-27:
在所述过渡元素化合物中,m是元素周期表中的第4族过渡金属,例如铪(hf)、锆(zr)、钛(ti)等,me是甲基,ph是苯基。
本发明涉及的催化剂组合物可以包含助催化剂化合物,所述助催化剂化合物可以包含选自所述过渡金属化合物以及由下述化学式3至化学式6来表示的化合物中的一种以上的化合物。
化学式3:
在所述化学式3中,al是铝,r27是卤素原子、碳原子数1至20的烃基或者碳原子数1至20的被卤素取代的烃基,a是2以上的整数,因此这是具有重复单元的化合物。
化学式4:
在所述化学式4中,al是铝或硼,r28是卤素原子、碳原子数1至20的烃基、碳原子数1至20的被卤素取代的烃基或者碳原子数1至20的烷氧基。
化学式5:
[l1-h]+[z1(a2)4]-
化学式6:
[l2]+[z2(a3)4]-
在所述化学式5及化学式6中,l1及l2是中性或阳离子路易斯酸,z1及z2是元素周期表中的第13族元素,a2及a3是被取代或未被取代的碳原子数6至20的芳基或者被取代或未被取代的碳原子数1至20的烷基
由所述化学式3表示的化合物为铝氧烷,只要是通常的烷基铝氧烷就没有特别的限定。例如,可以使用甲基铝氧烷、乙基铝氧烷、异丁基铝氧烷、丁基铝氧烷等,具体而言,可以使用甲基铝氧烷。可以通过向三烷基铝添加适量的水,或者使包含水的烃化合物或者无机水合物盐与三烷基铝发生反应等常规的方法制备所述烷基铝氧烷,一般得到的是直链状与环状的铝氧烷混合的形式。
例如,由所述化学式4表示的化合物可以使用常规的烷基金属化合物。具体而言,可以使用三甲基铝、三乙基铝、三异丁基铝、三丙基铝、三丁基铝、二甲基氯化铝、三异丙基铝、三环戊基铝、三戊基铝、三异戊基铝、三己基铝、三辛基铝、乙基二甲基铝、甲基二乙基铝、三苯基铝、三对甲苯基铝、二甲基甲氧基铝、二甲基乙氧基铝、三甲基硼、三乙基硼、三异丁基硼、三丙基硼、三丁基硼、三(五氟苯基)硼等,更具体而言,可以使用三甲基铝、三异丁基铝、三(五氟苯基)硼等。
例如,由所述化学式5或6表示的化合物可以使用甲基双十八烷基铵四(五氟苯基)硼酸盐([hnme(c18h37)2]+[b(c6f5)4]-)、三甲基铵四(苯基)硼酸盐,三乙基铵四(苯基)硼酸盐、三丙基铵四(苯基)硼酸盐、三丁基铵四(苯基)硼酸盐、三甲基铵四(对甲苯基)硼酸盐、三丙基铵四(对甲苯基)硼酸盐、三甲基铵四(o,p-二甲基苯基)硼酸盐、三乙基铵四(o,p-二甲苯基)硼酸盐、三甲基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(五氟苯基)硼酸盐、二乙基铵四(五氟苯基)硼酸盐、三苯基鏻四(苯基)硼酸盐、三甲基鏻四(苯基)硼酸盐、n,n-二乙基苯铵四(苯基)硼酸盐、n,n-二甲基苯铵四(五氟苯基)硼酸盐、n,n-二乙基苯铵四(五氟苯基)硼酸盐、三苯基碳鎓四(对三氟甲基苯基)硼酸盐、三苯基碳鎓四(五氟苯基)硼酸盐、三甲基铵四(苯基)铝酸盐、三乙基铵四(苯基)铝酸盐、三丙基铵四(苯基)铝酸盐、三丁基铵四(苯基)铝酸盐、三甲基铵四(对甲苯基)铝酸盐、三丙基铵四(对甲苯基)铝酸盐、三乙基铵四(o,p-二甲基苯基)铝酸盐、三丁基铵四(对三氟甲基苯基)铝酸盐、三甲基铵四(对三氟甲基苯基)铝酸盐、三丁基铵四(五氟苯基)铝酸盐、n,n-二乙基苯铵四(苯基)铝酸盐、n,n-二乙基苯铵四(苯基)铝酸盐、n,n-二乙基苯铵四(五氟苯基)铝酸盐、二乙基铵四(五氟苯基)铝酸盐、三苯基鏻四(苯基)铝酸盐、三甲基鏻四(苯基)铝酸盐、三乙基铵(苯基)铝酸盐、三丁基铵四(苯基)铝酸盐等,但是并不仅限于此。具体而言,可以使用甲基双十八烷基铵四(五氟苯基)硼酸盐([hnme(c18h37)2]+[b(c6f5)4]-)、n,n-二甲基苯铵四(五氟苯基)硼酸盐、三苯基碳鎓四(五氟苯基)硼酸盐等。
在制备本发明涉及的负载型混杂茂金属催化剂时,所述第一及第二茂金属化合物的过渡金属(所述化学式中1的m1及所述化学式2中的m2)与载体的质量比优选为1:1至1:1000。优选地,可以是1:100至1:500。当包含上述质量比的载体及茂金属化合物时,表现出适当的负载型催化剂活性,从而在催化剂活性的保持以及经济性方面有利。
此外,由化学式5、6代表的助催化剂化合物与载体的质量比优选为1:20至20:1,化学式3、4的助催化剂化合物与载体的质量比优选为1:100至100:1。
所述第一茂金属化合物与所述第二茂金属化合物的质量比优选为1:100至100:1。当包含上述质量比的助催化剂以及茂金属化合物时,在催化剂活性的保持以及经济性方面有利。
适于制备本发明涉及的负载型混杂茂金属催化剂的载体可以是具有大表面积的多孔性物质。
所述第一及第二茂金属化合物以及助催化剂化合物可以是被载体混杂负载而用作催化剂的负载型催化剂。负载型催化剂是指,被载体负载,以便良好地分散且稳定地保持,从而提升催化剂的活性并保持稳定性的催化剂。
混杂负载并非是指第一及第二茂金属化合物分别被载体负载,而是通过一次性过程使载体负载催化剂化合物。混杂负载缩短制备时间且减少溶剂使用量,因此可以说与分别负载相比更为经济。
所述载体是使具备催化剂功能的物质分散,稳定地负载并保持的固体,通常是多孔性或者面积大的物质,以便高度分散并负载催化剂功能物质,从而使其露出表面积变大。载体在机械、热、化学性方面需要稳定,载体的示例包括二氧化硅、氧化铝、氧化钛、沸石、氧化锌淀粉、合成聚合物等,但并不局限于此。
所述载体的平均粒度可以是10至250微米,优选为10至150微米,更优选为20至100微米。
所述载体的微孔体积可以是0.1至10cc/g,优选为0.5至5cc/g,更优选为1.0至3.0cc/g。
此外,所述载体的比表面积可以是1至1000m2/g,优选为100至800m2/g,更优选为200至600m2/g。
当所述载体为二氧化硅时,二氧化硅的干燥温度可以是200至900℃。优选为300至800℃,更优选为400至700℃。当低于200℃时,水分过多,导致表面的水分与助催化剂发生反应,而当超过900℃时,载体的结构崩溃。干燥后的二氧化硅内的羟基的浓度可以是0.1至5mmol/g,优选为0.7至4mmol/g,更优选为1.0至2mmol/g。当低于0.5mmol/g时,助催化剂的负载量低,而当超过5mmol/g时,催化剂成分惰性化,因此并非优选。
本发明涉及的负载型混杂茂金属催化剂可以通过使茂金属催化剂活化的步骤、使载体负载活化后的茂金属催化剂的步骤来制备。在制备所述负载型混杂茂金属催化剂时,可以首先使载体负载助催化剂。所述茂金属催化剂的活化可以分别进行,也可以根据情况而改变。即可以混合第一茂金属化合物与第二茂金属化合物并使其活化,然后使载体负载,可以先使载体负载助催化剂化合物之后再负载第一、第二茂金属化合物。
制备所述负载型混杂茂金属催化剂时的反应溶剂可以使用诸如己烷、戊烷的脂肪烃溶剂;诸如甲苯、苯的芳香烃溶剂;如二氯甲烷的被氯原子取代的烃溶剂;诸如二乙醚、四氢呋喃的醚类溶剂;丙酮、乙酸乙酯等大部分的有机溶剂,优选为甲苯、己烷,但并不仅限于此。
制备所述催化剂时的反应温度为0至100℃,优选为25至70℃,但并不仅限于此。
制备所述催化剂时的反应时间为3分钟至48小时,优选为5分钟至24小时,但并不仅限于此。
可以通过与所述助催化剂化合物混合(接触),来实现所述茂金属化合物的活化。所述混合通常可以在氮气或氩气的惰性环境下,不使用溶剂或者在所述烃溶剂存在的情况下进行。
所述第一及第二茂金属化合物的活化时的温度可以为0至100℃,优选为10至30℃。
当通过助催化剂活化所述第一及第二茂金属化合物时,搅拌时间可以是5分钟至24小时,优选为30分钟至3小时。
关于所述茂金属化合物,可以直接使用均匀溶解于所述烃溶剂等中的溶液状态的催化剂组合物,或者使用去除溶剂的固体粉末状态的,但并不仅限于此。
本发明涉及的高密度乙烯类聚合物的制备方法包括使所述负载型混杂茂金属催化剂与一种以上的烯烃单体接触,从而制备聚烯烃均聚物或者共聚物的步骤。
本发明的高密度乙烯类聚合物的制备方法(聚合反应)可以是淤浆相或者气相聚合反应。此外,可以根据聚合方法(淤浆聚合、气相聚合)所需的聚合结果或者聚合物的形态来多样地变更各个聚合反应条件。本领域技术人员容易地实施其变形程度。
当在液相或淤浆相下实施所述聚合时,可以将溶剂或者烯烃本身用作介质。所述溶剂的示例可以有丙烷、丁烷、戊烷、己烷、辛烷、癸烷、十二烷、环戊烷、甲基环戊烷、环己烷、甲基环己烷、苯、甲苯、二甲苯、二氯甲烷、氯乙烷、二氯乙烷、氯苯等,可以以规定的比例混合这些溶剂来使用,但是并不仅限于此。
在具体例中,所述烯烃单体的示例可以有乙烯、α-烯烃类、环烯烃类、二烯类、三烯(trienes)类、苯乙烯(styrenes)类等,但是并不仅限于此。
所述α-烯烃类包括碳原子数3至12,例如3至8的脂肪族烯烃,具体示例可以有丙烯、1-丁烯、1-戊烯、3-甲基-1-丁烯、1-己烯、4-甲基-1-戊烯、3-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯(1-decene)、1-十一烯、1-十二烯、1-十四烯、1-十六烯、1-二十烯、4,4-二甲基-1-戊烯、4,4-二乙基-1-己烯以及3,4-二甲基-1-己烯等。
所述α-烯烃类可以均聚,或者两种以上的交替(alternating)、随机(random)或嵌段(block)共聚。所述α-烯烃类的共聚包括乙烯与碳原子数3至12,例如3至8的α-烯烃的共聚(具体而言,乙烯和丙烯、乙烯和1-丁烯、乙烯和1-己烯、乙烯和4-甲基-1-戊烯、乙烯和1-辛烯等)以及丙烯与碳原子数4至12,例如碳原子数4至8的α-烯烃的共聚(具体而言,丙烯和1-丁烯、丙烯和4-甲基-1-戊烯、丙烯和4-甲基-1-丁烯、丙烯和1-己烯,丙烯和1-辛烯等)。在所述乙烯或丙烯与其它α-烯烃的共聚物中,其它α-烯烃的量可以是整体单体的99摩尔%以下,如果是乙烯共聚物,则优选为80摩尔%以下。
所述烯烃单体的优选示例有乙烯、丙烯、1-丁烯、1-己烯、1-辛烯、1-癸烯以及它们的混合物,但是并不仅限于此。
本发明的高密度乙烯类聚合物制备方法中,所述催化剂组合物的使用量并不特别的限制,但是举例的话,聚合的反应系统内由所述化学式1和2来表示的过渡金属化合物的中心金属(m,第4族过渡金属)的浓度可以是1×10-5至9×10-5mol/l。此外,聚合时的温度及压力会基于反应物质、反应条件等而发生变化,因此没有特别的限制,但如果是溶液聚合,则聚合温度可以是0至200℃,优选为100至180℃,如果是淤浆或气相聚合,则聚合温度可以是0至120℃,优选为60至100℃。此外,聚合压力可以是1至150巴(bar),优选为30至90巴,更优选为10至20巴。可以通过注入烯烃单体气体(例如,乙烯气体)来产生压力。
例如,所述聚合可以是间歇式、半连续式或者连续式。所述聚合也可以通过具有不同反应条件的两个以上的步骤来进行,可以通过改变聚合温度或者向反应器内注入氢气的方法来调节最终聚合物的分子量。
将所述负载型混杂茂金属化合物用作催化剂,通过乙烯均聚或者乙烯与α-烯烃的共聚来获得本发明涉及的高密度乙烯类聚合物,其具有单一分子量分布。
下面,对本发明涉及的高密度乙烯类聚合物进行具体的说明。
本发明的高密度乙烯类聚合物的密度可以是0.910至0.970g/cm3,更优选为0.950至0.965g/cm3。当聚合物的密度在0.940g/cm3以下时,无法具有足够的强韧性。当聚合物的密度在0.970g/cm3以上时,结晶度过大,成型体易于发生脆性破坏,因此并非优选。
一般,mi(熔融指数)变大时成型性提高,但是耐冲击性会降低。由于这种理由,当为了提高成型性而提高mi时,通常使用通过共聚来形成短链支化结构(降低密度),从而防止耐冲击性变差的方法。但是这种乙烯密度的降低导致聚合物的强韧性变差,因此在基于降低密度的应用方面,存在局限性。当降低mi时,耐冲击性以及耐化学性提高,但是熔融流动性变差,因而大幅降低成型性。不同于以往的高密度乙烯类聚合物,本发明的高密度乙烯类聚合物具有低mi,因此表现出优异的耐冲击性以及耐化学性。此外,具有宽分子量分布和长支链,因此表现出优异的注射成型性。
本发明中所说的熔融流动性主要对应于从挤出机挤出熔融树脂时的挤出负荷,成为这种熔融流动性的标准的指标为mi、mfi、mfr等。本发明中mi(熔融指数)表示190℃、2.16kg荷重下的流动性,mfi表示190℃、21.6kg荷重下的流动性。mfr表示mfi与mi之比,即mfi/mi。
本发明的高密度乙烯类聚合物的mi可以是0.1至50g/10min,优选为0.5至10g/10min。如果mi为0.1g/10min以下,则在用作注射成型材料时的成型加工性大幅降低,注塑产品的外观出现不良。如果mi大于50g/10min,则耐冲击性大幅降低。
本发明的高密度乙烯类聚合物的mfr可以是35至100,更优选为37至80。如果mfr为35以下,则在用作注射成型材料时的成型加工性大幅降低。如果mfr为100以上,则机械物理性质降低。
此外,本发明的高密度聚乙烯聚合物的特征在于,具有满足下述式1的mfr。
式1:
-10.11in(mi)+51.744>mfr>-12.25in(mi)+65.457
本发明的高密度乙烯类聚合物的悬臂梁(izod)冲击强度可以是4至30kj/m2,更优选为5至28kj/m2。如果悬臂梁冲击强度在4kj/m2以下,则对成型品的强度产生不利影响,如果悬臂梁冲击强度在30kj/m2以上,则对成型品的加工性产生不利影响。
本发明的高密度乙烯聚合物的特征在于,具有满足下述式2的悬臂梁冲击强度。
式2:
93.655in(mfr)–332.52>悬臂梁冲击强度>39.395in(mfr)+140.74
其中,悬臂梁冲击强度是测定冲击强度的方法之一,是固定试样的一面并竖起来后击打上部,由此测定强度的方法,对欲知冲击强度的材料施加冲击而破坏,测定由此产生的吸收能量,求出冲击强度,此外能够了解对于冲击的耐久性(durability)。
本发明的负载型混杂茂金属催化剂表现出优异的催化剂的活性,当利用本发明的负载型混杂茂金属催化剂制备烯烃聚合物时,可制备具有宽分子量分布,并且共聚单体集中于高分子量体中的聚合物。所述烯烃聚合物具有优异的冲击强度、弯曲强度、耐应力开裂性、熔融张力,因此可应用于吹塑成型体、注射成型体、薄膜、管、挤出成型体。
图1是为确认实施例3、4、8中所制备的共聚物的共聚单体分布而分析gpc-ir的曲线图,图2是为确认比较例5、8中所制备的共聚物的共聚单体分布而分析gpc-ir的曲线图。
已确认,本发明的高密度聚乙烯共聚物中高分子量体包含大量共聚单体,因此耐应力开裂性和冲击强度优异。一般,mi变低时能够提升机械物理性质,但是熔融流动性变差,使得加工性变差。但是已经确认,本发明的高密度乙烯类聚合物具有低mi而具有优异的机械物理性质,并且包含长支链,表现出高mfr,因此加工性优异。
关于本发明的流变性,在伸长率1(1/s)、2.5亨奇应变(henckystrain)(变形率)下,与下述比较例明显不同的应力(stress)(基于变形率的应力)可以是600000至1000000dyn/cm2。
图3比较了实施例4中制备的共聚物与商业产品基于压力的注射成型性。在200~220℃下10秒内向模具投入了聚合物,填充的试样长度越长,就表示加工性越优异。
图4比较了实施例1、2、3、4、6、7、8中制备的共聚物与商业产品基于mi的加工性。在200~220℃下10秒内以21巴的压力投入了聚合物,填充的试样长度越长,就表示加工性越优异。
图5比较了实施例1、2、3、4、6、7、8中制备的共聚物与商业产品基于mi的冲击强度。
图6比较了实施例1、4中制备的共聚物与商业产品的加工性。一般为了提高加工性,提高mi或者加宽分子量分布。如此即降低加工时的树脂年度,从而降低挤出注射时形成在螺旋桨上的负荷,因此可实现高速生产。但是当提高mi或者加宽分子量分布时,高分子聚合物的机械强度降低。然而参照图6即可确认,与现有的商业产品相比,本发明的高密度乙烯类共聚物在保持机械强度等物理性质的同时形成低负荷,因此可实现高速生产。
图7比较了实施例4中制备的共聚物与商业产品的流变性。图7中基于亨奇应变的应力值越高,就表示存在越多的长支链。一般的特征为,含有长支链时冲击强度变差。但是参照图5与下述表1、2即可确认,本发明的高密度乙烯类聚合物即使包含长支链,也表现出优异的冲击强度。
流变性是与物质的流动和变形有关的物理性质,在生产产品的工艺中,物质所经历的流动和变形,对产品的特征起到决定性的影响,当物质在经历流动和变形时表现的独特的性质就是该物质的流变学性。流变性的测定方法为,测定施加变形(strain)而出现的应力(stress),得到物质函数。
图7中增加着变形率的单位即亨奇应变,并以曲线图来表示基于此的应力,在伸长率1(1/s)、2.5亨奇应变下,与比较例明显不同的本发明的实施例4的应力为670000dyn/cm2。
实施例
下面,通过本发明的优选实施例,对本发明的构成以及作用进行更为详细的说明。只是,这些只是作为本发明的优选示例而提供,应解释为本发明在任何方面都不被其限制。
未在此记载的内容是本领域技术人员足以进行技术类推的,因此省略其说明。
第一茂金属化合物制备例1.(茚基(环戊二烯基))zrcl2
将茚(5g,0.043mol)溶于己烷(150ml)之后充分地混合,冷却至-30℃,然后向己烷溶液缓慢地滴落2.5m的正丁基锂(n-buli)己烷溶液(17ml,0.043mol),并在常温下搅拌12小时。用玻璃过滤器过滤白色悬浊液,再使白色固体充分干燥之后,制得茚锂盐(收率:99%)。
将茚锂盐(1.05g,8.53mmol)淤浆溶液和cpzrcl3(2.24g,8.53mmol)溶于乙醚(30ml)中后冷却至-30℃。向该乙醚溶液缓慢滴落溶于乙醚(15ml)的茚锂盐后搅拌24小时,制得(茚基(环戊二烯基))zrcl2([indenyl(cyclopentadienyl)]zrcl2)(收率:97%)。在此,cp表示环戊二烯基(cyclopentadienyl)。
第一茂金属化合物制备例2.(2-甲基苯并茚基(环戊二烯基))zrcl2
使用2-甲基苯并茚(2-methylbenzeindene),通过与制备例1相同的方法制得了(2-甲基苯并茚基(环戊二烯基))zrcl2([2-methylbenzeindenyl(cyclopentadienyl)]zrcl2)(收率:95%)。
第一茂金属化合物制备例3.(2-苯基苯并茚基(四甲基环戊二烯基))zrcl2
使用2-甲基苯并茚(2-methylbenzeindene)和四甲基环戊二烯基(tetramethylcyclopentadienyl),通过与制备例1相同的方法制得了(2-苯基苯并茚基(四甲基环戊二烯基))zrcl2([2-phethylbenzeindenyl(tetramethylcyclopentadienyl)]zrcl2)(收率:93%)。
第一茂金属化合物制备例4.(芴基(环戊二烯基))zrcl2
使用芴(fluorene)和环戊二烯(cyclopentadiene),通过与制备例1相同的方法制得了(芴基(环戊二烯基))zrcl2(收率:92%)。
第二茂金属化合物制备例5.me2si(2-甲基-4,5-苯并茚基)2zrcl2
制备例5-1:配体化合物的制备
将2-甲基苯并茚(2-methylbenzeindene)(2.13g,1eq)添加到30ml的己烷(haxane)中,并在-30℃下缓慢添加正丁基锂(n-buli)(8.1ml,1.1eq,1.6minhaxane)之后,将温度缓慢提升至常温并搅拌12小时。对生成的固体进行过滤,并用己烷洗涤之后,在真空下进行干燥,由此制得了浅灰色固体化合物。
对于所述灰色固体化合物,将5ml的sime2cl2(520mg,1eq)乙醚(ether)溶液中缓慢添加15ml的乙醚分散液之后慢慢将温度提升至常温(25℃),并搅拌12小时。利用乙醚与水的混合溶液,萃取有机层并进行干燥后用己烷洗涤,制得了((2-甲基苯并茚基)2sime2((2-methylbenzoindenyl)2sime2)(收率55%)。
制备例5-2:第二茂金属化合物的制备
将制备例5-1的化合物(0.4g,1eq)添加到15ml的thf(tetrahydrofuran,四氢呋喃)中,并在-30℃下缓慢添加正丁基锂(1.32ml,2.2eq,1.6minhaxane)之后,慢慢将温度提升至常温并搅拌12小时,由此制得二锂盐(dilithiumsalt),然后向二锂盐(dilithiumsalt)淤浆溶液缓慢地添加zrcl4(435mg,1eq)之后搅拌12小时。用真空去除溶剂,然后用thf、mc洗涤,从而制得了me2si(2-甲基-4,5-苯并茚基)2zrcl2(me2si(2-methyl-4,5-benzoindenyl)2zrcl2)(收率37%)。
第二茂金属化合物制备例6.me2si(2-甲基-4-(1-萘基))2zrcl2(me2si{2-methyl-4-(1-naphthyl)}2zrcl2)
制备例6-1:配体化合物的制备
将2-甲基-4-溴茚(2-methyl-4-bromoindene)(2g,1eq)、pd(pph3)4(553mg,0.05eq)、1-naphb(oh)2(2.14g,1.3eq)添加到thf、meoh溶液(4:1,40ml)中,然后在常温下注入经抽气(degassing)的k2co3水溶液(2.0m,3.3eq)后,在80℃下环流搅拌12小时,从而制得了2-甲基-4-(1-萘基)茚(2-methyl-4-(1-naphthyl)indene)。将2-甲基-4-(1-萘基)茚添加到50ml的甲苯中后,在-30℃下缓慢添加正丁基锂(7.8ml,1.1eq,1.6minhaxane)之后,将温度缓慢提升至常温并搅拌12小时。对生成的固体进行过滤,并用己烷洗涤之后,在真空下进行干燥,由此制得了2-甲基-4-(1-萘基)茚基锂(2-methyl-4-(1-naphthyl)indenyllithium)。
在-30℃下缓慢将sime2cl2(462mg,1eq)添加至13ml的2-甲基-4-(1-萘基)茚基锂(1.88g,2eq)甲苯、3ml的thf中后,缓慢提升温度,在55℃下搅拌12小时,从而制得了1.97g(97%)的二甲基双(2-甲基-4-(1-萘基)茚基)硅烷(dimethylbis{2-methyl-4-(1-naphthyl)indenyl}silane)。
制备例6-2:第二茂金属化合物的制备
利用二甲基硅烷基双(2-甲基-4-(1-萘基)茚基)二锂(dimethylsilylbis{2-methyl-4-(1-naphthyl)indenyl}dilithium),通过与制备例5-2相同的方法制得了me2si(2-甲基-4-(1-萘基))2zrcl2(me2si(2-methyl-4-(1-naphthyl))2zrcl2)(收率94%)。
第二茂金属化合物制备例7.me2si(2-甲基-4-(2-萘基))2zrcl2(me2si{2-methyl-4-(2-naphthyl)}2zrcl2)
制备例7-1:配体化合物的制备
利用2-甲基-7-(2-萘基)茚(2-methyl-7-(2-naphthyl)indene),通过与制备例5-1相同的方法制得了二甲基双(2-甲基-4-(2-萘基)茚基)硅烷(dimethylsilylbis{2-methyl-4-(2-naphthyl)indenyl}silane)(收率51%)。
制备例7-2:第二茂金属化合物的制备
利用制备例7-1的化合物,通过与制备例5-2相同的方法制得了me2si(2-甲基-4-(2-萘基))2zrcl2(me2si(2-methyl-4-(2-naphthyl))2zrcl2)(收率90%)。
第二茂金属化合物制备例8.me2si(2-甲基-4-苯基茚基)2zrcl2(me2si(2-methyl-4-phenylindenyl)2zrcl2)
制备例8-1:配体化合物的制备
将2-甲基-4-溴茚(2-methyl-4-bromoindene)(7g,1eq)、ni(dppp)cl2(363mg,0.02eq)添加到乙醚(ether)(100ml)中,在0℃下,将phmgbr3.0minether(13.3g,1.05eq)添加1小时。将温度缓慢提升至常温(25℃)后,在0℃下进行12小时的环流搅拌。反应结束后将溶液浸于冰浴(icebath)中,然后添加1nhcl酸,从而将ph降至4。用分液漏斗萃取有机层后,进行mgso4处理,从而除水。过滤并干燥溶剂,从而制得了,2-甲基-4-(苯基)茚(2-methyl-4-(phenyl)indene)(收率97%)。利用2-甲基-4-(苯基)茚,通过与制备例5-1相同的方法制得了me2si(2-甲基-4-苯基茚基)2(me2si(2-methyl-4-phenylindenyl)2)(收率95%)。在此dppp表示1,3-双(二苯基膦)丙烷(1,3-bis(diphenylphosphino)propane)。
制备例8-2:第二茂金属化合物的制备
利用me2si(2-甲基-4-苯基茚)2(me2si(2-methyl-4-phenylindene)2),通过与制备例5-2相同的方法制得了me2si(2-methyl-4-phenylindenyl))2zrcl2(收率90%)。
负载型混杂茂金属催化剂制备例9
第一及第二茂金属化合物和助催化剂即甲基铝氧烷(mao)与空气中的水分或氧气发生反应时会失去活性,因此所有的实验都采用手套箱、史兰克(schlenk)技术,在氮气条件下进行。对于10l负载催化剂反应器,进行洗涤以去除异物,并在110℃下干燥3小时以上,密封反应器之后利用真空完全地去除水分等,在此状态下进行使用。
向2.862g的制备例1化合物、3.469g的制备例8-2化合物添加甲基铝氧烷(mao)溶液(甲基铝氧烷:1188g),并在常温下搅拌1小时。将300g的二氧化硅(xpo2402)投入反应器中后,向反应器添加精制的甲苯900ml并进行搅拌。1小时的搅拌步骤结束之后,搅拌着反应器投入了第一茂金属化合物、第二茂金属化合物以及甲基铝氧烷混合溶液。将反应器升温至60℃之后,搅拌2小时。
沉淀反应之后去除上清液,并用甲苯1l进行洗涤之后,在60℃下真空干燥12小时。
负载型混杂茂金属催化剂制备例10
除了使用2.382g的制备例2化合物、4.387g的制备例8-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例11
除了使用2.712g的制备例3化合物、3.046g的制备例7-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例12
除了使用2.662g的制备例4化合物、3.712g的制备例6-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例13
除了使用2.359g的制备例1化合物、4.357g的制备例5-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例14
除了使用2.329g的制备例4化合物、4.357g的制备例8-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例15
除了使用2.359g的制备例1化合物、4.057g的制备例7-2化合物以外,通过与制备例9的方法进行制备。
负载型混杂茂金属催化剂制备例16
除了使用2.359g的制备例2化合物、4.157g的制备例5-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例17
除了使用2.159g的制备例2化合物、3.357g的制备例6-2化合物以外,通过与制备例9相同的方法进行制备。
负载型混杂茂金属催化剂制备例18
除了使用2.659g的制备例3化合物、4.557g的制备例5-2化合物以外,通过与制备例9相同的方法进行制备。
催化剂比较例1
除了使用制备例1化合物和(nbucp)2zrcl2以外,通过与制备例9相同的方法进行制备。
催化剂比较例2
除了使用(nbucp)2zrcl2和制备例8-2化合物以外,通过与制备例9相同的方法进行制备。
催化剂比较例3
除了使用制备例1化合物和me2si(me4cp)(ntbu)ticl2以外,通过与制备例9相同的方法进行制备。
催化剂比较例4
除了使用制备例8-2化合物以外,通过与制备例9相同的方法进行制备。
实施例1
将在所述制备例9中制得的负载型混杂茂金属催化剂投入到流化床气体工艺(fluidizedbedgasprocess)连续聚合器中,从而制备了烯烃聚合物。将1-己烯用作共聚单体,反应器乙烯压力保持为15巴,聚合温度保持为80~90℃。
实施例2至10
除了分别使用制备例10至18的负载型混杂茂金属催化剂以外,通过与实施例1相同的方法制备了烯烃聚合物。
比较例5至8
除了分别使用比较例1至4的负载型混杂茂金属催化剂以外,通过与实施例1相同的方法制备了烯烃聚合物。
<物理性质测定方法>
1)按照astm1505测定了密度。
2)mi、mfi以及mfr
熔融流动性mi是2.16kg荷重下10分钟内的挤出量,在测定温度190℃下按照astm1238进行了测定。
mfi是21.6kg荷重下10分钟内的挤出量,在测定温度190℃下按照astm1238进行了测定。
mfr表示mfi与mi之比,即mfi/mi。
3)分子量以及分子量分布(pdi)
利用凝胶渗透色谱法-傅氏转换红外线光谱分析仪(gpc-ftir)测定了平均分子量。
4)悬臂梁冲击强度
按照astmd256,测定了冲击强度。
5)耐环境应力开裂(escr)
按照astmd1693标准,将10个试样(sample)添加到50℃、10%的igepal溶液中后,以在五个试样上发生failure(crack)时为基准,评价了耐环境应力开裂(environmentalstresscrackingresistance,以下称escr)。
6)bocdi(broadorthogonalcomonomerdistributionindex,宽正交共聚单体分布指数)
显示scb(shortchainbranch,短支链)分布的数值,其值为正时,scb集中在高分子量体中。
7)树脂成型性评价(模具流动性)
按照astmd790标准,注射成型弯曲试样,从而评价加工性。
<注射条件>
料筒(barrel)温度:200至220℃,模具温度:30℃,注射压力:18巴、21巴,填充时间:10s,冷却时间:20s,压紧(packing):0巴弯曲试样缓冲长度:12cm。
在此,astm是标准的名称,分为如下5种:1)相应领域中的通用术语的定义,2)认为适于达成给定任务的顺序,3)用于进行给定测定的方法,4)对对象物或者概念进行分组的基准,5)规定产品或材料的特性的范围或界限等。
此外,mi即熔融指数表示塑料在规定荷重、规定温度下所具有的熔融流动性,该熔融指数大就意味着高分子的加工性优异,其与分子量成反比关系。聚烯烃类树脂有多种成型方法,但是这些方法的共同之处在于,首先加热树脂以形成熔融状态,并对此进行成型。因此在聚烯烃类树脂的成型加工方面熔融特性是极为重要的物理性质。特别是对于挤出、压缩、注射、旋转成型等成型来说,熔融特性即熔融流动性是左右满意的成型性的根本性的物理性质。熔融流动指数越大,就意味着越易于流动。
在本发明中,mi表示190℃、2.16kg荷重下的流动性,mfi表示190℃、21.6kg荷重下的流动性。mfr表示mfi与mi之比,即mfi/mi。
此外,悬臂梁冲击强度是测定冲击强度的方法之一,是固定试样的一面并竖起来后击打上部,由此测定强度的方法,对欲知冲击强度的材料施加冲击而破坏,测定由此产生的吸收能量,求出冲击强度,此外能够了解对于冲击的耐久性(durability)。
此外,根据成型条件或者保管及运送时的温度、时间、环境等,高分子材料的物理性质受到敏感的影响,特别是至今也都未能准确地预测长时间的物理性质变化,因此也有可能出现意料之外的破损。在疲劳(fatigue)或蠕变(creep)环境下,在相对低的应力状态下发生破损,这与一般的金属材料相同,但是高分子材料接触化学熔剂时有可能在极低的应力或变形率状态下也发生开裂。如此因外部刺激而发生的环境应力开裂(environmentalstresscracking)现象是包含熔剂的吸收及渗透、混合物的热力学、空蚀现象(cavitation)、部分材料的屈服现象等的复杂的现象。特别是,被报告的高分子材料的破损原因中,环境应力开裂所引起的比值达到15至20%,因此耐环境应力开裂性成为高分子材料的重要数值。
本发明的物理性质测定中,通过上述的方法进行耐环境应力开裂性的测定。
与此同时,本发明的物理性质测定中测定bocd指数。bocd是最近开发的新型概念的高分子结构相关术语,bocd结构是指,如ɑ-烯烃的共聚单体的含量集中于高分子量主链中的结构,即越趋向高分子量侧,短链结构的含量就越多的结构。
以重均分子量(mw)为基准,在分子量分布(mwd)左右30%范围内,测定scb(shortchainbranching,短支链)含量(单位:个/1000c),再通过下述式3求值,由此求出bocd指数。
式3
可以认为,若bocd指数在0以下,那就不是bocd结构的高分子,而若大于0,就是bocd结构的高分子,其值越大,bocd特性就越优异。
在表1中整理了所述实施例1至10以及比较例6至8的聚合条件。
表1
下面的表2中示出了上述的物理性质测定数据(data)。此外,已经确认,与以往进行商用的9种hdpe(高密度聚乙烯)相比,本发明实施例的高密度乙烯类聚合物具有优异的熔融流动性以及优异的机械物理性质。
已确认,本发明的高密度乙烯类聚合物中的高分子量体中包含较多的短支链,因此表现出优异的耐应力开裂性和冲击强度特性。一般降低mi时,能够提升机械物理性质,但是熔融流动性变差,使得加工性变差。已确认,本发明的高密度乙烯类聚合物具有低mi,因此机械物理性质优异,并且由于含有长支链,表现出高mfr,因此加工性优异。本发明的高密度乙烯类聚合物虽然具有低mi,但是由于高mfr特性,加工性表现优于现有的hdpe。所使用的商业产品为,比较例9的a产品(hdpe7303、密度0.9523g/cm3、mi2.1g/10min)、比较例10的b产品(hdpe9100、密度0.9556g/cm3、mi2.4g/10min)、比较例11的c产品(hdpeme2500、密度0.9538g/cm3、mi2.1g/10min)、比较例12的d产品(hdpem850、密度0.9642g/cm3、mi4.9g/10min)、比较例13的e产品(hdpe2200j、密度0.9582g/cm3、mi5.1g/10min)、比较例14的f产品(hdpe7210、密度0.9581g/cm3、mi6.1g/10min)、比较例15的g产品(hdpem680、密度0.9557g/cm3、mi6.9g/10min)、比较例16的h产品(hdpem2210j、密度0.9582g/cm3、mi8.0g/10min)、比较例17的i产品(hdpeme8000、密度0.9592g/cm3、mi8.0g/10min)。
表2
从图1、图2和上述的表1及表2可确认,与比较例相比,将化学式1的第一茂金属和化学式2的第二茂金属混杂负载的茂金属催化剂表现出更高的活性,因而经济性更加优异。此外,表现出高mfr,因此注射加工性优异,并且bocdi具有正值,因此确认了短支链多分布于高分子量体中。
此外,已经确认,因为如上所述的理由,利用本发明的混杂茂金属催化剂制备的高密度乙烯类聚合物的冲击强度和耐应力开裂性优异。
此外参照图3和图4、表1和表2,本发明的高密度乙烯类聚合物虽具有低mi,但具有高mfr特性,因此表现出优于现有的hdpe的加工性。
并且如图2所示,关于escr,可确认出现5个开裂的时间如下:当乙烯类聚合物的密度为0.950至0.960g/cm3且mi为1.0至3g/10min时,为16小时以上至100小时以下;当所述密度为0.950至0.965g/cm3且所述mi为6.0至7.0g/10min时,为10小时以上至80小时以下;当所述密度为0.955至0.960g/cm3且所述mi为8.0至9.0g/10min时,为9小时以上至75小时以下
在制备负载型混杂茂金属时,与具有对称结构的比较例7相比,使用本发明的具有不对称结构的化学式1的第一茂金属时表现出更加优异的活性。
不对称结构中,由于从配体将电子传递至中心金属的电子传递现象并不相同,中心金属与配体之间的结合长度会有不同,因此单体接近催化剂活性点时所遇到的立体障碍小。然而,在对称结构中,由于向中心金属传递相同的电子,中心金属与配体之间的结合长度相同,因此立体障碍大于不对称结构中的立体障碍,所以在形成高密度聚合物时表现出低活性。
第二茂金属结构不同于化学式2的比较例6中,虽然表现出优异的活性,但是加工性差,高分子量体少,共聚单体集中于低分子量体中,因此熔融张力和耐应力开裂性低,难以进行商用。化学式2的第二茂金属具有交联结构形式,因此保护催化剂活性点,并且使共聚单体易于接近催化剂活性点,因而共聚单体的侵入性优异。此外与非交联结构相比,催化剂活性点稳定,因此能够形成高分子量。但是化学式2的茂金属单独存在时,活性过低,并不经济,并且产生过多的高分子量体,使得加工性变差。
以上例示了本发明的优选实施例,但是本发明并不局限于上述的特定的优选实施例,当然,本领域技术人员都可以在不脱离权利要求书中所记载的本发明的主旨的范围内实施各种变形,这样的变形都落在权利要求书的记载范围之内。
- 还没有人留言评论。精彩留言会获得点赞!