抑制畸胎瘤形成的多能干细胞衍生的神经前体细胞的制备方法与流程
本发明涉及一种从多能干细胞抑制畸胎瘤形成的神经前体细胞的制备方法。
背景技术:
多能干细胞是一种能够在保持未分化状态的同时无限增殖并且在一定的环境和条件下可以分化成特定功能和形态的细胞。人类多能干细胞可在适当的培养条件下无限增殖(自我更新;self-renewal),并且,可分化为构成个体的各种细胞。因此,针对理解对个体发育、分化及生长的基本知识和开发用于治疗个体损伤或疾病的治疗剂,而正在进行各方面的研究。
胚胎干细胞作为多能干细胞中的一种,与细胞分裂停止的分化细胞不同,可以进行自我更新而无限地生成与自我更新细胞相同的细胞,并且,具有可通过多种环境或刺激分化为身体的所有功能细胞的分化全能性。因此,当细胞或器官受损时,可将胚胎干细胞用作其分化的特定细胞,进而,利用胚胎干细胞的疗法正成为各种难治愈性疾病的基本治疗方法。
人诱导性多能干细胞(inducedpluripotentstemcells,ipsc)作为多能干细胞的类型之一,是指将已分化的细胞通过遗传修饰或者利用外分泌因子的重编程而进行逆分化的细胞,使细胞具有分化全能性。ipsc也可以与胚胎干细胞类似地进行自我更新,并可分化为身体的所有类型的细胞。迄今为止,诱导性多能干细胞在基因表达和分化潜能中,具有与胚胎干细胞几乎相同的特征。
含有神经前体细胞而从人类多能干细胞分化的特定分化细胞利用在特定分化细胞中被表达的各个固有表面标志(surfacemarker),可通过流式细胞分选法(fluorescenceactivatedcellsorting,facs)或磁性激活细胞分选法(magneticactivatedcellsorting,macs)分离。但是,在磁性激活细胞分选法的情况下,纯度相对低且没有高回收率,并且在一些情况下,可导致根据磁性材料的变形。在流式细胞分选法的情况下,具有激光损坏细胞的缺点。并且,当分离来自多能干细胞的分化细胞时,由于磁性激活细胞分选法及流式细胞分选法都具有未分化的多能干细胞的混淆可能性,因此,仅分离具有100%纯度的分化细胞受到技术性限制。因此,在利用多能干细胞而开发的细胞治疗剂中,不能排除畸胎瘤形成的可能性。因此,开发一种从已分化的细胞群选择性去除未分化多能细胞的技术,其不影响分化细胞且导致畸胎瘤发生的风险,对于多能干细胞衍生细胞治疗剂的临床和商业化是必不可少的。
并且,在对作为人类多能干细胞转录因子的oct-4、nanog及sox-2等与作为细胞表面标志的tra-1-60、tra-1-81、ssea3及ssea4等的抗体从分化细胞群分离及去除多能干细胞时使用阴性选择(negativeselection)方法。但是,据报道,由于大多数上述因子或标志具有碳水化合物表位或者其功能对于人类多能干细胞的自我再生产或分化全能性维持不是必需的,因此,不能保证准确地和选择性地仅分离未分化多能干细胞。并且,存在通过基因操作使在特定时期的未分化细胞自然死亡的方法,但是这些基因操作方法具有其效率低并且其安全性未得到验证的缺点。
技术实现要素:
技术问题
为此,本发明人在为了在多能干细胞衍生神经前体细胞的制备中找出抑制畸胎瘤形成的方法而进行研究的过程中,确认了本发明的畸胎瘤抑制方法在培养过程中自然去除未分化多能干细胞且阻断畸胎瘤发生的可能性,从而完成了本发明。
本发明的目的在于,提供一种抑制畸胎瘤形成的多能干细胞衍生的神经前体细胞的制备方法。
解决问题的手段
为了实现上述目的,本发明提供一种抑制畸胎瘤形成的神经前体细胞的制备方法,包括:对神经前体细胞进行单细胞化的步骤;对上述单细胞化的神经前体细胞进行自发性再凝集的步骤;以及对上述再凝集的神经前体细胞进行回收的步骤。
并且,本发明提供通过上述抑制畸胎瘤形成的神经前体细胞的制备方法制备而成的神经前体细胞。
与此同时,本发明提供包括分化上述神经前体细胞的步骤的神经细胞的分化方法。
发明的效果
根据本发明的神经前体细胞的制备方法不仅提高神经前体细胞的纯度并抑制畸胎瘤的形成,而且增加细胞的存活率及回收率。
附图说明
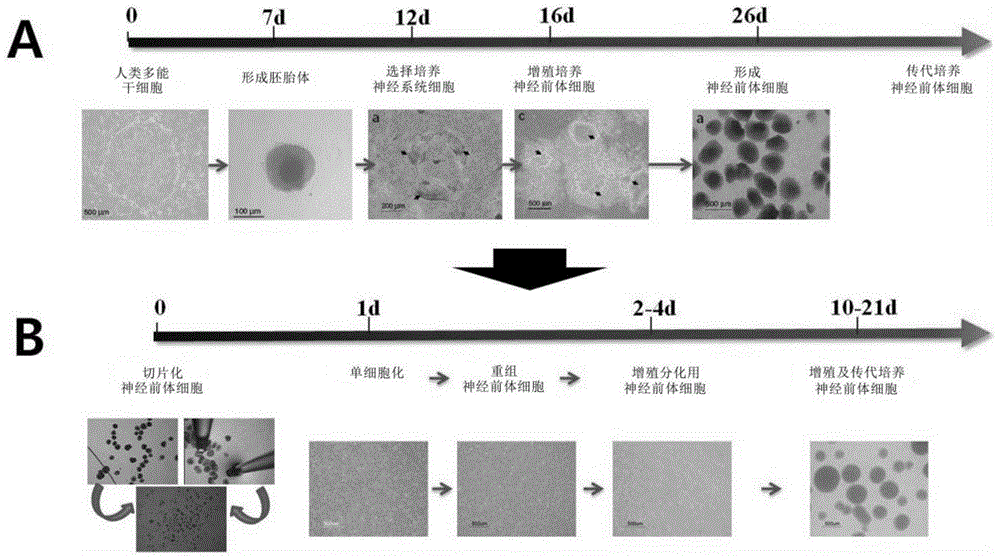
图1为对本发明的神经前体细胞的制备方法(b)与现有方法(a)进行比较的图。
图2为通过神经细胞标志的表达与否来确认神经前体细胞分化为神经细胞的分化能力的图。
图3为通过免疫染色来确认在神经前体细胞中神经前体细胞标志的表达与否的图。
图4为确认细胞(a)及细胞(b)的存活率的图表,上述细胞(a)为对神经前体细胞进行作为常规方法的酶处理来进行单细胞化的细胞,上述细胞(b)为切片化后进行粘附培养而酶处理的细胞。
图5为示出在神经前体细胞被单细胞化后进行自发性凝集的过程中在培养皿中出现的多个凝集物的性状及大小变化的图。
图6为通过流式细胞分析来确认未分化多能干细胞标志(a)及神经前体细胞标志(b)在胚胎神经前体细胞(snm)及再凝集的神经前体细胞(reform)中的表达的曲线图。
图7为通过rt-pcr来确认未分化标志及神经前体细胞标志在再凝集的神经前体细胞中的表达与否的图。
图8为通过免疫组织学染色来确认再凝集的神经前体细胞的分化能力是否维持的图。
具体实施方式
以下,详细说明本发明。
本发明提供一种抑制畸胎瘤形成的神经前体细胞的制备方法,包括:步骤1),对神经前体细胞进行单细胞化;步骤2),对在上述步骤1)的单细胞化的神经前体细胞进行再凝集;以及步骤3),对在上述步骤2)的再凝集的神经前体细胞进行回收。本发明的制备方法简要示出在图1的(b)部分,以往的制备方法简要示出在图1的(a)部分。
本说明书中所使用的术语“神经前体细胞”意味着从诸如人体胚胎干细胞或诱导性多能干细胞的多能干细胞形成的神经前体细胞或者从神经组织分离的神经前体细胞以球的形态凝集。
在上述制备方法中,可将加入n-2添加剂、bfgf或其的混合物的培养基用作细胞的培养用培养基。
本说明书中所使用的术语“n-2添加剂(supplement)”作为基于bottenstein’sn-1制剂(formulation)而化学定义的无血清补充剂,在周围神经系统与中枢神经系统的一次培养分裂之后,为了神经细胞及神经母细胞瘤的生长与表达,可一同加入到补充有bfgf及egf等的生长因子的培养用培养基中来使用。
相对于神经前体细胞培养用培养基的总体积,上述n-2添加剂的添加量为0.5%(v/v)至2.0%(v/v),更具体地,可以为1.0%(v/v)至1.5%(v/v)。若上述n-2添加剂的添加量小于0.5%(v/v),则细胞的生长效率可能会降低,若大于2.0%(v/v),则过度营养的增殖可对细胞培养的维持产生影响。根据本发明的一实施例,相对于神经前体细胞培养用培养基的总体积,上述n-2添加剂的添加量可以为1%(v/v)。
本说明书中所使用的术语“bfgf(碱性成纤维细胞生长因子;basicfibroblastgrowthfactor)”意味着对正常成纤维细胞及确立的细胞株均强有力的有丝分裂刺激因子。上述bfgf可具有细胞的移动、新型血管的形成、神经细胞的维持、内分泌功能的调节、特定细胞性蛋白的表达、细胞外基质的生产及细胞生存的促进等的功能。
相对于神经前体细胞培养用培养基的总质量,上述bfgf的添加量为0.0002%(w/v)至0.001%(w/v),更具体地,可以为0.0006%(w/v)至0.0008%(w/v)。若上述bfgf的添加量小于0.0002%(w/v),则细胞的生长及分裂效率可能会降低,若大于0.001%(w/v),则过度营养的增殖可对细胞的培养产生影响。根据本发明的一实施例,相对于神经前体细胞培养用培养基的总质量,上述bfgf的添加量可以为0.00063%(w/v)。
上述步骤1)及步骤3)可在涂敷有底物的培养皿中进行。上述底物可以为选自由层粘连蛋白、l-鸟氨酸、基质胶及cellstart组成的组中的一种以上。在本发明的一实施例中,上述底物可以为基质胶。
为了再凝集神经前体细胞,神经前体细胞之间凝集的力量应强于神经前体细胞要粘附到培养皿的力量。因此,与步骤1)及步骤3)不同,在步骤2)中,可通过使用未涂敷有底物的培养皿来诱导神经前体细胞之间的凝集。上述步骤1)的单细胞化可包括使用显微切片工具来对神经前体细胞进行的切片化。在此情况下,当使用细胞分离酶进行切片化时,由于细胞受损而回收率降低,因此可使用物理方法。作为切片化工具,可以使用能够在解剖显微镜下切直径为500μm以下的凝集物的任何工具,具体地,可以使用精细拔出的玻璃巴斯德移液管、直径为100μm以下的金属线及具有微米尺寸的孔的金属网等。根据本发明的一实施例,可通过精细拔出的玻璃巴斯德移液管来进行上述切片化。
并且,可以使用如胰蛋白酶-edta、分散酶(dispase)、accutase及胶原酶等本领域熟知的酶作为细胞分离用酶来对上述切片化的神经前体细胞进行单一化。根据本发明的一实施例,可以使用accutase。
上述步骤2)的培养可在未涂敷有底物的培养皿中进行。并且,若在涂敷有底物的培养皿中进行培养,则固定的神经前体细胞可分化为神经细胞。但是,若使用未涂敷有底物的培养皿,则不分化为神经细胞且神经前体细胞可自发性移动而神经前体细胞之间可再凝集。
上述步骤3)的再凝集的神经前体细胞的直径为10μm至80μm,具体地,可以为20μm至60μm。根据本发明的一实施例,上述神经前体细胞的直径可以为20μm至50μm。当上述再凝集的神经前体细胞的直径为约10μm至80μm时,可通过移液管吸取(pipetting)方式从培养皿的底部移除来传代。在此情况下,传代的方式为以48小时的间隔更换培养基的悬浮培养方式。
当从底部移除神经前体细胞时,可一同移除不凝集而残留在底部上的神经前体细胞。反之,当更换培养基时,可一同移除不沉淀在底部而悬浮的神经前体细胞。神经前体细胞具有培养皿或神经前体细胞之间不断连续粘附的特征。因此,可将以6小时至12小时的间隔从培养皿的底部移除再凝集的神经前体细胞的过程重复进行。进而,可预先防止神经前体细胞之间的过度结合。在悬浮培养期间,新形成的神经前体细胞以球形的形态增殖,当尺寸为适于目的的尺寸时,可回收使用。
在本发明的具体实施例中,本发明人证实,当本发明的神经前体细胞在分化为神经前体细胞标志以及神经细胞时,表达神经细胞标志(参照图2及图3)。并且,当对神经前体细胞切片化后,进行单细胞化时,神经前体细胞的细胞存活率及回收率大大增加(参照图4),而且,未分化多能干细胞标志在再凝集的神经前体细胞中几乎不表达。反之,确认了神经前体细胞标志在再凝集的神经前体细胞中强烈表达(参照图6)。并且,确认了在再凝集的神经前体细胞中,未分化标志不进行表达,但神经前体细胞标志进行表达(参照图7)。而且,通过免疫组织学染色确认了在粘附培养的神经前体细胞中神经细胞标志的表达(参照图8)。
因此,本发明的神经前体细胞的制备方法可有效地用于制备抑制畸胎瘤形成的神经前体细胞。
并且,本发明提供一种通过本发明的方法制备而成的神经前体细胞。
上述神经前体细胞可根据如上所述的制备方法制备而成。具体地,上述方法可包括:步骤1),对神经前体细胞进行单细胞化;步骤2),对在上述步骤1)的单细胞化的神经前体细胞进行再凝集;以及步骤3),对在上述步骤2)的再凝集的神经前体细胞进行回收。
在上述制备方法中,可将加入n-2添加剂、bfgf或其的混合物的培养基用作细胞的培养用培养基。相对于神经前体细胞培养用培养基的总体积,上述n-2添加剂的添加量为0.5%(v/v)至2.0%(v/v),更具体地,可以为1.0%(v/v)至1.5%(v/v)。根据本发明的一实施例,相对于神经前体细胞培养用培养基的总体积,上述n-2添加剂的添加量可以为1%(v/v)。并且,相对于神经前体细胞培养用培养基的总质量,上述bfgf的添加量为0.0002%(w/v)至0.001%(w/v),更具体地,可以为0.0006%(w/v)至0.0008%(w/v)。根据本发明的一实施例,相对于神经前体细胞培养用培养基的总质量,上述bfgf的添加量可以为0.00063%(w/v)。
上述步骤1)及步骤3)的培养可在涂敷有底物的培养皿中进行,上述底物可以为选自由层粘连蛋白、l-鸟氨酸、基质胶及cellstart组成的组中的一种以上。在本发明的一实施例中,上述底物可以为基质胶。
在上述步骤2)中,可通过使用未涂敷有底物的培养皿来诱导神经前体细胞之间的凝集。若使用涂敷有底物的培养皿,则不分化为神经细胞且神经前体细胞可自发性移动而神经前体细胞之间可再凝集。并且,上述步骤1)的单细胞化可包括使用显微切片工具来对神经前体细胞进行的切片化。在此情况下,当使用细胞分离酶进行切片化时,由于细胞受损而回收率降低,因此可使用物理方法。作为切片化工具,可以使用能够在解剖显微镜下切直径为500μm以下的凝集物的任何工具,具体地,可以使用精细拔出的玻璃巴斯德移液管、直径为100μm以下的金属线及具有微米尺寸的孔的金属网等。根据本发明的一实施例,可通过精细拔出的玻璃巴斯德移液管来进行上述切片化。
并且,可以使用如胰蛋白酶-edta、分散酶(dispase)、accutase及胶原酶等本领域熟知的酶作为细胞分离用酶来对上述切片化的神经前体细胞进行单一化。根据本发明的一实施例,可以使用accutase。同时,上述步骤3)的再凝集的神经前体细胞的直径为10μm至80μm,具体地,可以为20μm至60μm。根据本发明的一实施例,上述神经前体细胞的直径可以为20μm至50μm。
在本发明的具体实施例中,本发明人对神经前体细胞进行单细胞化,并通过在未涂敷有底物的培养基中对单细胞化的神经前体细胞进行培养来制备抑制畸胎瘤形成的神经前体细胞。并且,确认了上述制备的神经前体细胞在分化为神经前体细胞标志以及神经细胞时,表达神经细胞标志(参照图2及图3)。并且,当对切片化的神经前体细胞进行单细胞化时,神经前体细胞的细胞存活率及回收率增加(参照图4),而且,未分化多能干细胞标志在再凝集的神经前体细胞中几乎不表达。反之,确认了神经前体细胞标志在再凝集的神经前体细胞中强烈表达(参照图6)。并且,确认了在再凝集的神经前体细胞中,未分化标志不进行表达,神经前体细胞标志进行表达(参照图7)。
同时,本发明提供包括分化上述神经前体细胞的步骤的神经细胞的分化方法。
上述神经前体细胞可包括如上所述的特征。并且,在上述分化中,代替bfgf来加入的b27的添加量为总分化用培养基的1%(v/v)至3%(v/v),更具体地,可以为1.5%(v/v)至2.5%(v/v)。若上述b27的添加量小于1%(v/v),则细胞的分化效率可能会降低,若大于3%(v/v),则过度分化及增殖可对细胞的培养产生影响。根据本发明的一实施例,上述b27的添加量可以为2%(v/v)。
上述神经前体细胞能够以400μm至600μm的直径进行传代。上述神经前体细胞在直径小于400μm时进行传代的情况下,神经前体细胞的增殖可能会延迟,在600μm以上的直径进行传代的情况下,营养供应不顺畅而可导致神经前体细胞的内部细胞凋亡。根据本发明的一实施例,若上述再凝集的神经前体细胞的直径达到500μm,则将其重新切片化来进行传代培养或者可冷冻保存。
在本发明的一具体实施例中,本发明人在符合神经细胞的分化条件的情况下,通过涂敷有神经分化底物的培养皿和加入b27来代替bfgf的培养基,来将再凝集的神经前体细胞分化为神经细胞。在上述分化的细胞中,通过免疫组织学染色来确认神经细胞标志的表达(参照图8)。
发明的实施方式
以下,通过以下实施例详细说明本发明。
然而,以下实施例仅是例示本发明,本发明的内容并不限定于此。
<实施例1>神经前体细胞的培养准备
根据本发明,通过神经前体细胞重组过程,从由多能干细胞形成的神经前体细胞的多个切片中仅纯分离出神经前体细胞。
<1-1>用于培养神经前体细胞的培养基的制备
首先,通过在490ml的dmem(f12)加入5ml的0.1mm的非必需氨基酸(nonessentialaminoacids,neaa)和5ml的2mmp/s(青霉素(penicillin)/链霉素(streptomycin))来制备神经前体细胞的基础培养基。将制备的培养基充分混合后,使用孔径为0.22μm的过滤器并用真空泵过滤,并在每50ml的管中分配49ml。将上述管用石蜡膜密封并在4℃的温度下进行保存。若培养基的量少于12ml,则置于15ml的管中保存,若培养基的量在12ml以上,则置于50ml的管中保存。
另一方面,通过在49.5ml的上述所制备的基础培养基加入0.5ml的1%的n-2添加剂(supplement)和0.4ml的80μg/ml的bfgf(碱性成纤维细胞生长因子;basicfibroblastgrowthfactor)来制备神经前体细胞的培养用培养基。
<1-2>用于培养神经前体细胞的培养皿的准备
用于培养神经前体细胞的培养皿的尺寸根据神经前体细胞的数量决定。在本实施例中,每35mm直径的培养皿使用10个切片化的神经前体细胞(1个凝集物/cm2)。在使用培养皿之前,将1ml的稀释20倍的基质胶加入培养皿中,在室温下保存1小时以上。
<实施例2>神经前体细胞的确认
<2-1>确认神经前体细胞分化为神经细胞的分化能力
首先,通过对将在实验中所使用的神经前体细胞进行免疫染色来确认上述细胞是否具有分化为神经细胞分化能力。
从人体胚胎干细胞株(snuhes3)及诱导性多能干细胞株(f5ips,y2-3ips)制备神经前体细胞,即如下。培养7天后,将增殖的人体胚胎干细胞株(snuhes3)及诱导性多能干细胞株(f5ips,y2-3ips)机械分成每个具有200个至300个细胞的块,然后在支持细胞上分别重新进行培养。接着,将2mg/ml的胶原酶(collagenaseⅳ;invitrogen,carlsbad,ca,usa)在37℃的温度下处理40分钟后移除干细胞集群(colony),然后将其加入到微生物培养用培养皿,在去除bfgf的干细胞培养基中悬浮培养7天至21天来制备胚状体(embryoidbody,eb)。在胚状体中除去囊肿形态的囊性胚状体(cysticeb)后,将剩余的胚状体粘附到基质胶(
并且,为了免疫染色实验,使用精细拔出的玻璃巴斯德移液管的工具来将神经前体细胞切片化后,在涂敷有分化底物的玻璃盖玻片上进行粘附培养。所使用的培养用培养基为加入有n2与b27的神经细胞分化用培养基。培养2周后,确认了是否能观察到对作为神经细胞的标志的betaiii-tubuline(bⅲ-tub)和作为经分化的神经细胞的芽细胞群的多巴胺神经细胞的标志的酪氨酸羟化酶(tyrosinehydroxylase,th)进行表达的细胞。将样品在3%的福尔马林溶液处理20分钟后,保存到磷酸缓冲液中。在处理抗体之前,将样品用封闭液(2%的牛血清白蛋白(bovineserumalbumin),sigma)处理一天,与第一抗体反应12小时,与第二抗体反应1小时,用含有dapi的荧光保存用溶液密封之后,在共聚焦荧光显微镜(nikon,japan)下进行观察。在此情况下,为了免疫学染色,将rabbitanti-betaiiitubulinantibody(1:100,chemicon)和mouseanti-th(1:200,chemicon)用作第一抗体,alexafluor488donkeyanti-mouseigg和alexafluor594donkeyanti-rabbitigg(1:200,molecularprobes,usa)用作第二抗体。
结果,如图2所示,作为多巴胺神经细胞的标志的酪氨酸羟化酶(tyrosinehydroxylase,th)及作为神经细胞的标志的bⅲ-tub(beta3tubuline)在神经前体细胞中表达(图2)。
<2-2>确认在神经前体细胞的神经前体细胞标志的表达
并且,通过免疫染色来确认上述神经前体细胞是否表达神经前体细胞的标志。神经前体细胞在冷冻切片化后,粘附在盖玻片上进行免疫染色,免疫染色方法在与实施例<2-1>相同的条件和方法下进行。在此情况下,为了免疫学染色,将rabbitanti-musashiantibody(1:100,chemicon)、rabbitanti-sox1antibody(1:200,milipore)、mouseanti-humannestinantibody(1:200,chemicon)及mouseanti-pax6antibody(1:500,novusbiologicals)用作第一抗体,alexafluor488donkeyanti-mouseigg及alexafluor594donkeyanti-rabbitigg(1:200,molecularprobes,usa)用作第二抗体来进行试验。
结果,如图3所示,作为神经前体细胞标志的巢蛋白、musashi及pax6在神经前体细胞中表达(图3)。
<实施例3>神经前体细胞的制备
<3-1>神经前体细胞的切片化(schizolysis)
首先,使用显微切片工具将神经前体细胞以不超过100μm3的体积的尺寸进行切片化。具体地,使用精细拔出的玻璃巴斯德移液管(pipette)来对神经前体细胞进行切片化。使用<实施例1>的神经前体细胞培养用培养基及涂敷基质胶的培养皿,在37℃的温度和5%的co2条件下对切片化的神经前体细胞进行12小时的粘附培养。
<3-2>确认切片化的神经前体细胞的单细胞化及细胞存活率
切片化的神经前体细胞的单细胞化
对切片化的神经前体细胞使用如下方法来进行单细胞化(细胞化)。
首先,去除在<3-1>中培养的神经前体细胞的培养基,用磷酸盐缓冲盐水(phosphatebufferedsaline,pbs)轻轻洗涤。去除pbs并加入0.2ml/cm2的accutase后,在37℃的温度和5%的co2条件下,反应40分钟至60分钟来对粘附的神经前体细胞的多个切片进行单细胞化。将对其进行离心分离并聚集后,去除accutase且用神经前体细胞培养用培养基洗涤,加入1ml的培养用培养基后计数。
根据单细胞化过程变化确认细胞存活率的增加
然后,确认了对神经前体细胞进行切片化并进行单细胞化的过程是否对细胞的存活率及回收率产生影响。
具体地,对单细胞化的细胞进行1天的粘附培养后,再次进行单细胞化,然后用台盼蓝(trypanblue)进行染色并用自动细胞计数器测定经染色的细胞的细胞数及存活率。并且,作为对照组,在相同条件下比较将切片化后立即进行酶处理而不进行粘附培养的细胞群。
结果,如图4所示,对神经前体细胞进行切片化并进行粘附培养后,进行单细胞化的神经前体细胞的细胞存活率及回收率大大增加。
<3-3>诱导单细胞化的细胞的自发性再凝集
在上述<3-2>中,为了将单细胞化的细胞再次自发性再凝集,进行了如下处理。
首先,将单细胞化的多个细胞以5.5×104cell/0.2ml/cm2的浓度稀释于神经前体细胞培养用培养基中,并在37℃的温度和5%的co2条件下,对其进行24小时的培养。在此情况下,为了使神经前体细胞不分化为神经细胞并进行自发性再凝集,在未涂敷有底物的培养皿中进行培养。24小时后,首次将凝集物移液管吸取后悬浮,然后通过用移液管吸取将凝集物从培养皿的底部移除的方式,以48小时间隔更换培养用培养基进行悬浮培养,直至细胞凝集物的直径最小为约20μm且不超过50μm。当从底部移除凝集物时,可一同移除不凝集而残留在底部上的多个细胞,并且更换培养基的同时一同移除仅把凝集物沉淀在底部且悬浮的细胞。由于多个凝集物保持粘附于培养皿或其他凝集物的特性,因此,对再凝集的多个凝集物以最少12小时的间距使用如上所述的方法来防止与培养皿或凝集物的过度结合。
结果,如图5所示,确认了细胞单细胞化后,随着培养时间再凝集形成的凝集物的性状及尺寸发生了变化。这意味着,可根据凝集物的尺寸在适当时期回收再凝集的神经前体细胞来用作分化(第4天)、传代培养或冷冻保存(第21天)等。
<3-4>再凝集的神经前体细胞的分化
当在上述<3-3>中再凝集的神经前体细胞的直径达到50μm至100μm时,将其回收并诱导向神经细胞的分化。
具体地,将再凝集的分化用神经前体细胞全部回收并悬浮在神经前体细胞培养用培养基中,然后移至2.5倍至3倍面积的培养皿或具有三个相等面积的培养皿以进行分化。在再凝集过程中,使用了6cm直径的培养皿,将再凝集的神经前体细胞全部回收并悬浮在15ml的培养用培养基后,移至75cm2的t-烧瓶进行分化。分化时,使用了如<实施例1>中涂敷基质胶的培养皿。培养24小时后,更换加入2%(v/v)的b27来代替bfgf的培养基来诱导向神经细胞的分化。若上述再凝集的神经前体细胞的直径达到500μm,则将其重新切片化来进行传代培养或者可冷冻保存。
<实验例1>确认再凝集的神经前体细胞的特性
确认了再凝集的神经前体细胞的多个细胞是否照依旧维持神经前体细胞的特性。
<1-1>流式细胞分析
首先,处理accutase来对神经前体细胞进行单细胞化后,将其用pbs洗涤并用固定液固定后进行免疫染色。通过对作为未分化多能干细胞的熟知标志的ssea-4及tra-61蛋白质的染色来确认未分化细胞是否存在。通过利用作为神经前体细胞的标志的musashi及pax6蛋白质来确认神经前体细胞的纯度。人体胚胎干细胞(es)用作对照组,并将以往的神经前体细胞群(snm)和重组的神经前体细胞群(reformedsnm)一同进行比较。
结果,如图6所示,主要在胚胎干细胞中表达的作为未分化多能干细胞标志的ssea-4及tra1-60蛋白质在再凝集的神经前体细胞中几乎不表达(图6的a部分)。反之,作为神经前体细胞的标志的musashi及pax6蛋白质在构成再凝集的神经前体细胞的大部分细胞中表达,并且,相比以往的snm,强烈表达神经前体细胞标志的细胞才能以单细胞群形态来表示(图6的b部分)。这些结果表明,再凝集的神经前体细胞不仅去除未分化细胞,也可纯分离到同一分化阶段的细胞。
<1-2>rt-pcr
通过rt-pcr来确认在人体胚胎干细胞、胚胎体、神经前体细胞及再凝集的神经前体细胞各个中的基因表达。基因的表达通过主要在未分化干细胞中表达的oct4及nanog,主要在神经前体细胞中表达的sox1、pax6及巢蛋白,以及在初期神经细胞中表达的nf-m的标志来确认。
具体地,使用trizol溶液(invitrogen)分别从人体胚胎干细胞、胚胎体、上述<实施例2>的神经前体细胞及上述<3-3>的再凝集的神经前体细胞中提取细胞的rna,使用cdna反转录试剂盒(accupowerrtpremix;bioneer)来合成cdna。以合成的cdna为模型,通过利用对oct4、nanog、sox1、pax6、巢蛋白(nestin)及nf-m的各个引物和taqpolymerase(hipiplus5xpcrpremix;elpisbiotech,taejeon,korea)来执行rt-pcr。rt-pcr中所使用的引物碱基序列如下列表1。
表1
结果,如图7所示,未分化标志在再凝集的神经前体细胞中不表达,而增加了多个神经前体细胞标志的表达。
<1-3>免疫组织学染色
为了确认再凝集的神经前体细胞的分化能力是否继续维持,执行了免疫组织学染色。
通过利用加热玻璃巴斯德移液管后精细拔出而制造的工具来对<3-3>的再凝集的神经前体细胞进行切片化后,在涂敷有作为与上述<实施例1>相同的底物的基质胶的玻璃盖玻片,以神经前体细胞培养用培养基进行1天,以神经细胞分化用培养基进行6天的粘附培养。培养第2天和第7天回收了粘附培养的玻璃盖玻片。样品在3%的福尔马林溶液处理20分钟后,保存到磷酸缓冲液。在处理抗体之前,将样品用封闭液(2%的牛血清白蛋白(bovineserumalbumin),sigma)处理一天,与第一抗体反应12小时,与第二抗体反应1小时,用含有dapi的荧光保存用溶液密封之后,在共聚焦荧光显微镜(nikon,japan)下进行观察。在此情况下,为了免疫学染色,将rabbitanti-betaiiitubulinantibody(1:100,chemicon)和mouseanti-humannestinantibody(1:200,chemicon)、mouseanti-thantibody(1:200,chemicon)及mouseanti-gabaantibody(1:200,chemicon)用作第一抗体,alexafluor488donkeyanti-mouseigg、alexafluor594donkeyanti-rabbitigg及alexafluor488donkeyanti-rabbitigg(以上1:200,molecularprobes,usa)用作第二抗体。
结果,如图8所示,与现有的神经前体细胞相同的作为经分化的神经细胞的标志的betaiii-tubuline、th及gaba蛋白质全部在粘附培养后分化7天的再凝集的神经前体细胞中表达。进而,确认了再凝集的神经前体细胞维持其分化能力。
序列表
<110>jeilpharmaceuticalco.,ltd
<120>preparingmethodsforpluripotentstemcellderived-neuronal
progenitorcellinhibitingformationofteratoma
<130>jeil1.20p
<150>kr10-2016-0184325
<151>2016-12-30
<160>14
<170>patentinversion3.2
<210>1
<211>20
<212>dna
<213>人工序列
<220>
<223>gapdhf
<400>1
accacagtccatgccatcac20
<210>2
<211>19
<212>dna
<213>人工序列
<220>
<223>gapdhr
<400>2
tccaccaccctgttgctgt19
<210>3
<211>22
<212>dna
<213>人工序列
<220>
<223>04-octf
<400>3
cgttctctttggaaaggtgttc22
<210>4
<211>20
<212>dna
<213>人工序列
<220>
<223>04-octr
<400>4
acactcggaccacgtctttc20
<210>5
<211>23
<212>dna
<213>人工序列
<220>
<223>nanogf
<400>5
gcgtacgcaaattaaagtccaga23
<210>6
<211>20
<212>dna
<213>人工序列
<220>
<223>nanogr
<400>6
tgctattcttcggccagttg20
<210>7
<211>20
<212>dna
<213>人工序列
<220>
<223>pax6f
<400>7
ggcaacctacgcaagatggc20
<210>8
<211>20
<212>dna
<213>人工序列
<220>
<223>pax6r
<400>8
tgagggctgtgtctgttcgg20
<210>9
<211>20
<212>dna
<213>人工序列
<220>
<223>sox1f
<400>9
caatgcggggaggagaagtc20
<210>10
<211>20
<212>dna
<213>人工序列
<220>
<223>sox1r
<400>10
ctctggaccaaactgtggcg20
<210>11
<211>21
<212>dna
<213>人工序列
<220>
<223>nestinf
<400>11
cagctggcgcacctcaagatg21
<210>12
<211>23
<212>dna
<213>人工序列
<220>
<223>nestinr
<400>12
agggaagttgggctcaggactgg23
<210>13
<211>22
<212>dna
<213>人工序列
<220>
<223>nf-mf
<400>13
gagcgcaaagactacctgaaga22
<210>14
<211>23
<212>dna
<213>人工序列
<220>
<223>nf-mr
<400>14
cagcgatttctatatccagagcc23
- 还没有人留言评论。精彩留言会获得点赞!