基于CRISPR/cas9和过氧化物酶APEX2系统识别分析特异性基因组位点相互作用DNA的方法与流程
本发明属生物
技术领域:
,具体涉及一种基于crispr/cas9和过氧化物酶apex2系统识别分析特异性基因组位点相互作用dna的方法。
背景技术:
:现有技术公开了在真核生物细胞中,dna分子具有高度组织性,并紧密地包裹着核小体的重复单元形成染色质。然而,在活细胞中,染色质的结构是动态变化的,局部的染色质可以被调控元件例如转录因子和非编码rna接近。近年来,本领域已经提出了若干调节染色质组织的机制,例如,真核细胞细胞核中的每一条染色体都位于一个被称为染色体区的特定区域,它包含许多通常大小为几百万碱基的结构域,称为拓扑结构域;在拓扑结构域中,远端dna元件通过动态相互作用来调控基因表达,研究显示,许多作用因子,包括ctcf、黏连蛋白和其他dna结合蛋白,都参与了拓扑结构域的形成和它们之间远程的相互作用。另外,表观遗传的修饰,例如dna的甲基化和组蛋白的修饰,还有长链非编码rnas都在通过调控染色质的高级结构来控制基因的表达过程中发挥着重要作用。这些发现使该
技术领域:
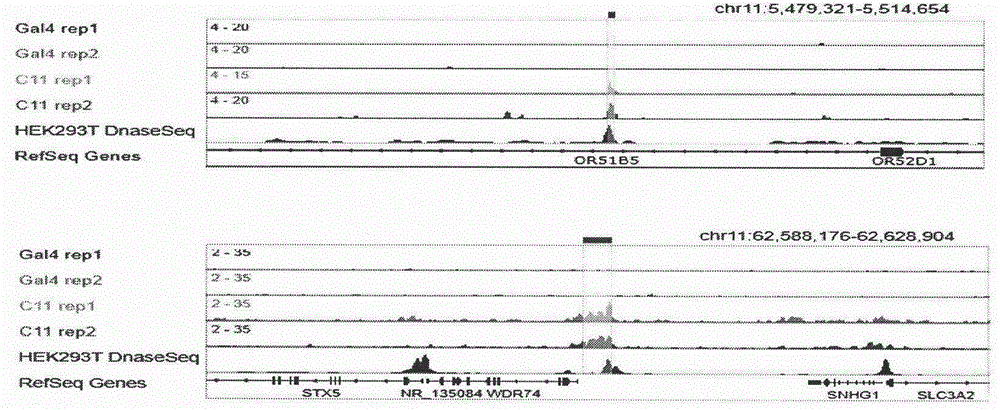
进入了一个染色质功能研究的新时代,然而,对染色质功能的全面了解需要识别存在于特定基因组位点的dna、rna及转录因子,该方面的研究由于技术上的困难而具有挑战性。近年来,有研究提出了局部染色质组成的技术,例如,染色质免疫共沉淀(chip)是其中的一种经典的,广泛用于研究给定蛋白全基因组分布的技术,然而,目前还没有一种被广泛接受的研究给定基因组位点局部相互作用分子的方法;锁定的核酸探针被用来识别与端粒区域结合的蛋白,但是这种方法仅限于高度重复的基因组区域;lexadna结合位点在基因层面上被整合到酵母基因组中,用于位点特异性的染色质纯化;然而,该方法需要对目标基因组进行基因组的改造,从而会改变染色质的自然环境,并且效率低;改良的基因组编辑技术,如转录激活剂类似的效应核酸酶(talen)和规律成簇的间隔短回文重复序列(crispr/cas9)已经被用来富集所需的基因组位点,然而,基于talen的方法要求为每个位点设计一段氨基酸序列,基于crispr的方法需要将细胞用甲醛交联,并且需要有高亲和性和特异性的抗体可用,等等;研究实践显示,上述的方法尚不能特异性地提供对天然的染色质或全基因组的功能分析。基于现有技术的研究基础,本申请的发明人拟提供一种识别分析特异性基因组位点相互作用dna的方法。尤其是一种识别分析任何给定染色体位置的局部相互作用dna的新方法。与本发明相关的现有技术有如下参考文献:1.j.andkingston,r.e.(2009)purificationofproteinsassociatedwithspecificgenomicloci.cell,136,175-86.2.byrum,s.d.,raman,a.,taverna,s.d.andtackett,a.j.(2012)chap-ms:amethodforidentificationofproteinsandhistoneposttranslationalmodificationsatasinglegenomiclocus.cellrep.,2,198-205.3.byrim,s.d.,taverna,s.d.andtackett,a.j.(2013)purificationofaspecificnativegenomiclocusforproteomicanalysis.nucleicacidsres.,41,e195.4.fujita,t.andfujii,h.(2013)efficientisolationofspecificgenomicregionsandidentificationofassociatedproteinsbyengineereddna-bindingmolecule-mediatedchromatinimmunoprecipitation(enchip)usingcrispr.biochem.biophys.res.commun.,439,132-6.5.waldrip,z.j.,byrum,s.d.,storey,a.j.,gao,j.,byrd,a.k.,mackintosh,s.g.,wahls,w.p.,taverna,s.d.,raney,k.d.andtackett,a.j.(2014)acrispr-basedapproachforproteomicanalysisofasinglegenomiclocus.epigenetics,9,1207-11.6.hung,v.,udeshi,n.d.,lam,s.s.,loh,k.h.,cox,k.j.,pedram,k.,carr,s.a.andting,a.y.(2016)spatiallyresolvedproteomicmappinginlivingcellswiththeengineeredperoxidaseapex2.nat.protoc.,11,456-75.技术实现要素:本发明的目的在于基于现有技术的研究基础,提供一种识别分析特异性基因组位点相互作用dna的方法,尤其是一种基于crispr/cas9和过氧化物酶apex2系统识别分析特异性基因组位点相互作用dna的方法。本方法操作简单、灵敏度高、特异性高、重现性好、适应于在任何给定的染色体位置上研究染色质局部相互作用dna的要求。基于本申请的前期研究基础,为了探究特定基因组位点相互作用的分子,提出一种称为caplocus(combiningcrisprandperoxidaseapex2systemtoidentifylocalchromatininteractions)的方法,并通过捕获人类染色体的端粒区,13号染色体一个重复区域和11号染色体一个单拷贝位点来验证该系统;通过全基因组测序分析,caplocus可以富集目标区域以及与之相互作用远距离基因组位点。本发明提出了一种基于crispr/cas9和过氧化物酶apex2系统识别分析特异性基因组位点相互作用dna的方法,该方法可识别分析任何给定染色体位置的局部相互作用dna。为实现上述目的,本发明采用如下的技术方案:1.载体构建,本发明中,使用的质粒系统包括三个质粒,分别表达dcas9、apex2和sgrna;2.细胞转化及生物素标记,本发明中,采用聚乙烯亚胺(pei)(polysciencesinc.,warrington,pa,usa)进行细胞的瞬时转染,细胞转染24h后,用500um的biotin-phenol(irisbiotechgmbh,germany)处理细胞30min,之后加入过氧化氢(h2o2)至终浓度1mm处理细胞1min之后,立即加入反应终止液(10mm叠氮化钠,10mmvc,5mmtrolox)终止反应,细胞用1%的甲醛固定10分钟,甘氨酸终止反应;3.将步骤2得到的细胞进行链霉素磁珠富集,富集得到的dna进行qpcr检测靶定区域的富集度并构建dna文库;4.将步骤3中检测合格的文库利用illumina平台hiseqxten测序仪进行测序;5.对测序结果进行peakcalling分析,获得与目标序列互作的基因组位点信息。本发明进行了实验分析,结果显示,该方法适用于任意基因组位点的分析,可获得与之互作的基因组位点信息,同时,该方法将crispr基因编辑系统与apex2结合进行特异位点dna分析的思路同样适用于其他的基因组编辑方法(如talen)与apex2的结合。本发明方法具有以下优点:1)caplocus可以有效地富集重复的基因组区域和高分辨率的单拷贝基因组位点;2)caplocus可以富集目标区域以及与之相互作用的远距离基因组位点。3)本方法操作简单、灵敏度高、特异性高、重现性好、适应于在任何给定的染色体位置上研究染色质局部相互作用dna的要求。为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图和具体实施例,对本发明进一步详细说明。应当理解,下述所描述的具体实施例仅用以解释本发明,并不用于限定本发明。以下实施例中所涉及的原料,如无特别说明均为市售,所涉及检测方法如无特别说明,则均为常规方法。下述的实施例是结合人类11号染色体单拷贝区(chr11:5,497,611-5,497,843)对本发明的方法进行进一步说明。附图说明图1,显示了(a)caplocus系统流程图及(b)相关质粒结构图示。图2,显示了caplocus系统特异性及富集效率图:其中,图a为caplocus系统靶向特定重复位点端粒区(telomere)的免疫荧光定位图;图b为dna富集qpcr定量结果图,其中ga14为阴性对照。图3显示了caplocus系统特异性捕获特定单拷贝位点(chr11:5,497,611-5,497,843)。图4显示了单拷贝位点chip-seq同时富集目标序列及与之互作位点。具体实施方式实施例1、质粒表达载体的构建和转染1.1质粒表达载体的构建:为了构建ms2-apex2_nls融合蛋白表达载体,用pcr的方法从pcdna3connexin43-gfp-apex2(addgeneplasmid:49385)质粒上扩增得到apex2,然后将其用bamhiandxhoi.酶切,克隆到phage-efs-mcp-3xbfpnls(addgeneplasmid:75384)载体上,通过将退火连接的寡聚核苷酸片段(oligos)和经bbsi酶切位点处连接到plh-sgrna1-2xms2(addgeneplasmid:75389)质粒上完成sgrna表达载体的构建,用addgene质粒64107表达dcas9;所有的sgrna序列和如表1所示;表1所有的sgrna序列namesequence(5’-3’)sgrna-telomeretagggttagggttagggttasgrna-gal4aacgactagttaggcgtgtasgrna-c20-1ctcttagctgttatgctgtasgrna-c20-2ggattcccttccatgctgaasgrna-c20-3ggtgcccaaaaaatagtcacsgrna-c20-4acactgaacaagccgataag1.2hek293t细胞培养:hek293t细胞在5%co2和37℃培养箱中培养,培养基为高葡萄糖dulbecco’smodifiedeagle’smedium(dmem;lifetechnologies,carlsbad,ca,usa),内加有浓度为10%的胎牛血清(sigma,st.louis,mo,usa),1%的青霉素/链霉素(lifetechnologies),每两天以1∶5的比例传代细胞,并检测一下支原体;1.3细胞转染:用聚乙烯亚胺(pei)(polysciencesinc.,warrington,pa,usa)根据厂家给的步骤进行细胞的瞬时转染,900ng的dcas9质粒,4.5ng的sgrna质粒和120ng的ms2-apex2_nls质粒共转到汇合度60%-80%的t75细胞培养瓶中,实施例2、提取dna进行chip实验2.1细胞转染24h后,当2x107个细胞被转染了目标sgrna或者gal4对照时,用500um的biotin-phenol(irisbiotechgmbh,germany)处理细胞30min,之后加入过氧化氢(h2o2)至终浓度1mm,处理细胞1min,之后,立即加入反应终止液(10mm叠氮化钠,10mmvc,5mmtrolox)终止反应,最后,用1%甲醛固定并在室温下孵育10min。用1ml的hypotonicbuffer(20mmhepesph7.5,10mm氯化钾,1mmedta,0.1mm激活的原巩酸钠,0.2%np-40,10%甘油)裂解细胞15min,细胞裂解物在4℃下,13000g离心1min,弃上清;2.2用500ulchipdilutionbuffer(20mmtris-hclph8.0,2mmedta,150mmnacl,0.1%sds,1%tritonx-100)重悬;2.3将重悬的沉淀在bioruptorpicosonicationdevice(非接触式超声波破碎仪)进行超声破碎,至片段长度在100bp-500bp之间;2.4在4℃离心机中,14000g,离心15min,之后将5%的上清取出作为input样品保存在-20℃;2.5用50ul的proteina在4℃将剪断的染色质预处理1h;2.6取出上清加到50μlm-280链霉亲和素免疫磁珠中,4℃旋转孵育过夜;2.7孵育过夜后,按顺序用2%sds,high-saltbuffer(50mmhepesph7.5,500mmnacl,1mmedta,0.1%sodiumdeoxycholate,1%tritonx-100),liclbuffer(10mmtris-hclph8.0,250mmlicl,1mmedta,0.5%np-40,0.5%sodiumdeoxycholate)andtris-edta(te)buffer,对磁珠进行清洗各一次;2.8将特异性结合了蛋白的磁珠和input样品加入sdselutionbuffer(50mmtris-hclph8.0,10mmedta,1%sds),70℃水浴孵育过夜进行反交联;2.9加入蛋白酶k和rna酶a处理溶液;2.10将2.9处理后的溶液用qiaquickpcrpurificationkit(qiagen,hilden,germany)纯化;2.11纯化的dna进行qrt-pcr的模板来检测每一个靶定区域的富集度;pcr的引物如表2所示。表2qpcr引物序列表c11-ip1-fcacacaagtgacaacaccagcc11-ip1-ratttaccctctgagtgccccc11-ip2-ftccatgctgaaggggtctgac11-ip2-rtaccaagtcacaaaggaggctc11-ip3-ftgagcctcctttgtgacttggc11-ip3-rtttgggttggtgacatcactgc11-1kbup-ftgcactttcccttcactgggc11-1kbup-rtcaggctaaggggttagggggc11-0.5kbup-fgcagcctcacagtaccatcttc11-0.5kbup-raggcatgaagagagggtttggc11-0.5kbdown-fctgaatttcgtgagcgccagc11-0.5kbdown-rgtgcacctctgtgctcatctc11-1kbdown-fgagcaacgcagaagacgggtc11-1kbdown-ractccctgaccccttgtgctc11-interact-ip1fcgacccaaggaagaaagggtc11-interact-ip1raggggatcttcagcctcgtc11-interact-ip2fgaggctgaagatcccctgtgc11-interact-ip2rgaaagcacgcagggataccac11-interact-0.5kbdown-ftttccagatcacagtagcccgc11-interact-0.5kbdown-rgtcctgggacaaaggataggctel-ip-fcggtttgtttgggtttgggtttgggtttgggtttgggtttel-ip-rggcttgccttacccttacccttacccttacccttaccct2.12qpcr反应体系为:2xsybrgreenqpcrmix10ul引物r1ul引物f1ul模板xul去离子水补足20ul;反应程序为:95℃10s;进入循环阶段:95℃变性10s,56℃退火30s,72℃延伸30s,共40个循环,每个循环在退火阶段采集荧光信号并在扩增结束后按同一条件分析数据,确定各样品的ct值;2.13根据2.12qpcr分析结果,纯化dna用ultraiidnalibraryprepkitforillumina(neb)构建dna文库,2.14建立的测序文库在hiseqxten仪器平台,根据厂商的说明进行测序。每一个文库的测序深度在11.74-35.83millionreads之间;2.13通过peakcalling软件对sgc11位点与sggal4阴性对照样品测序数据进行分析,重叠的峰用bedtools进行去除。从而确定了靶定位点和潜在的相互作用位点,用igv软件进行可视化。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1