一种环烷烃类化合物的合成方法与流程
本发明属于催化领域,具体涉及一种环烷烃类化合物的合成方法。
背景技术:
加氢反应在石油化工生产中具有广泛的应用,其中将芳烃通过催化加氢的方法获得饱和的环烷烃具有十分重要的意义。例如,通过催化加氢的方法将燃油中的芳烃转化为饱和环烷烃,可以提高燃油的安定性并减少芳烃不完全燃烧造成的环境污染,因此通过催化芳烃加氢得到环烷烃具有重要的经济价值和社会意义。
目前芳烃加氢适宜的催化剂主要是负载型贵金属催化剂、金属硫化物催化剂和过渡金属基催化剂,如铂(pt)、钯(pd)和铑(rh)等贵金属催化剂及镍(ni)基非贵金属催化剂。pt、pd贵金属催化剂具有催化活性高、反应条件温和等优点,在芳烃类化合物催化加氢生成环烷烃化合物占有很重要的地位。虽然贵金属有这些优点,但是贵金属催化剂价格成本过高,对原料杂质要求苛刻,且容易积碳。金属硫化物催化剂的寿命长,但活性低,操作条件苛刻,需要较高的温度和压力。负载型镍催化剂在制备过程中易发生团聚,催化效率较低,容易发生副反应。
由上述可知,开发在空气中稳定并具有优异催化性能的加氢还原催化剂用于芳烃类化合物的加氢还原,是本领域亟待解决的问题。
需注意的是,前述背景技术部分公开的信息仅用于加强对本发明的背景理解,因此它可以包括不构成对本领域普通技术人员已知的现有技术的信息。
技术实现要素:
本发明提供一种环烷烃类化合物的合成方法,该方法采用含核壳结构的碳包覆镍的纳米复合材料为催化剂,其丰富的介孔结构,有利于催化反应的传质,其包覆严密的金属纳米粒子,有利于在更苛刻的条件下发挥作用,用于芳烃类化合物加氢还原合成环烷烃类化合物,具有优异的活性、选择性及安全性。
为了实现上述目的,本发明采用如下技术方案:
本发明提供一种环烷烃类化合物的合成方法,包括:
以碳包覆镍的纳米复合材料为催化剂,所述催化剂与芳烃类化合物在氢气气氛下进行加氢还原反应;
其中,所述纳米复合材料含具有壳层和内核的核壳结构,所述壳层为掺杂氧的石墨化碳层,所述内核为镍纳米颗粒,其中所述纳米复合材料为具有至少一个介孔分布峰的介孔材料。
根据本发明的一个实施方式,其中所述芳烃类化合物的芳香环上还含有取代基,所述取代基选自c1-20的烷基、环烷基和芳基中的一种或多种。
根据本发明的一个实施方式,其中所述芳烃类化合物选自苯、甲苯。
根据本发明的一个实施方式,其中所述催化剂占所述芳烃类化合物质量的1-50%,优选为5-30%。
根据本发明的一个实施方式,其中所述加氢还原反应温度一般为200-300℃。
根据本发明的一个实施方式,其中所述氢气的压力为3-6mpa。
根据本发明的一个实施方式,所述加氢还原反应的反应时间优选为6~10h。
根据本发明的一个实施方式,其中所述催化剂与芳烃类化合物在溶剂中混合后进行加氢还原反应,其中所述溶剂选自醇类、醚类、烷烃类和水中的一种或多种。
根据本发明的一个实施方式,其中所述催化剂与醛类化合物在溶剂中混合后进行加氢还原反应,所述溶剂选自醇类、醚类、烷烃类和水中的一种或多种。
根据本发明的一个实施方式,上述合成方法中的纳米复合材料为具有至少一个介孔分布峰的介孔材料。任选地,所述纳米复合材料为具有两个或两个以上介孔分布峰的介孔材料。任选地,所述纳米复合材料在2~7nm的孔径范围和8~20nm的孔径范围分别具有一个介孔分布峰。任选地,其中所述介孔材料中介孔体积占总孔体积的比例大于50%,优选大于80%。任选地,所述纳米复合材料的介孔体积为0.05-1.25cm3/g,介孔体积也可以为0.30-0.50cm3/g。任选地,所述石墨化碳层的厚度为0.3nm~6.0nm,优选为0.3~3nm。任选地,所述核壳结构的粒径为1~200nm,优选为3~100nm,更优选为4~50nm。任选地,以质量百分比计,所述纳米复合材料的碳含量为10-60%,镍含量为30.0%-85.0%,优选地,碳含量为15.0%-40.0%,镍含量为50.0%-80.0%。任选地,其中以质量百分比计,该纳米复合材料中,氧含量小于15.0%,优选为0.2%-5.0%。上述纳米复合材料中各组分的含量之和为100%。
根据本发明的一个实施方式,上述合成方法中的纳米复合材料的酸洗损失率≤10%。任选地,所述纳米复合材料为具有至少一个介孔分布峰的介孔材料。任选地,所述纳米复合材料为具有两个或两个以上介孔分布峰的介孔材料。任选地,所述纳米复合材料在2~7nm的孔径范围和8~20nm的孔径范围分别具有一个介孔分布峰。任选地,其中所述介孔材料中介孔体积占总孔体积的比例大于50%,优选大于80%。任选地,所述纳米复合材料的介孔体积为0.05-1.25cm3/g,介孔体积也可以为0.30-0.50cm3/g。任选地,所述石墨化碳层的厚度为0.3nm~6.0nm,优选为0.3~3nm。任选地,所述核壳结构的粒径为1~200nm,优选为3~100nm,更优选为4~50nm。任选地,以质量百分比计,所述纳米复合材料的碳含量为15%-60%,镍含量为30%-80%,优选地,碳含量为30%-60%,镍含量为30%-60%。任选地,以质量百分比计,该纳米复合材料中,氧含量小于15.0%,优选为1.0%-10.0%。上述纳米复合材料中各组分的含量之和为100%。
本发明的有益效果在于,
本发明提供的环烷烃类化合物的合成方法,以碳包覆镍的纳米复合材料作为催化剂对芳烃类化合物进行加氢还原,由于催化剂材料非常稳定,不自燃,抗氧化,耐酸腐蚀,危险性低,适合保存与运输,从而保证了环烷烃类化合物合成过程的安全性。
本发明的碳包覆镍的纳米复合材料在催化还原芳烃类化合物为环烷烃类化合物的反应中表现了良好的重复性、高活性及高选择性,由于该纳米复合材料具有较强的磁性,还可方便利用其磁性分离催化剂或用于磁稳定床等工艺。
附图说明
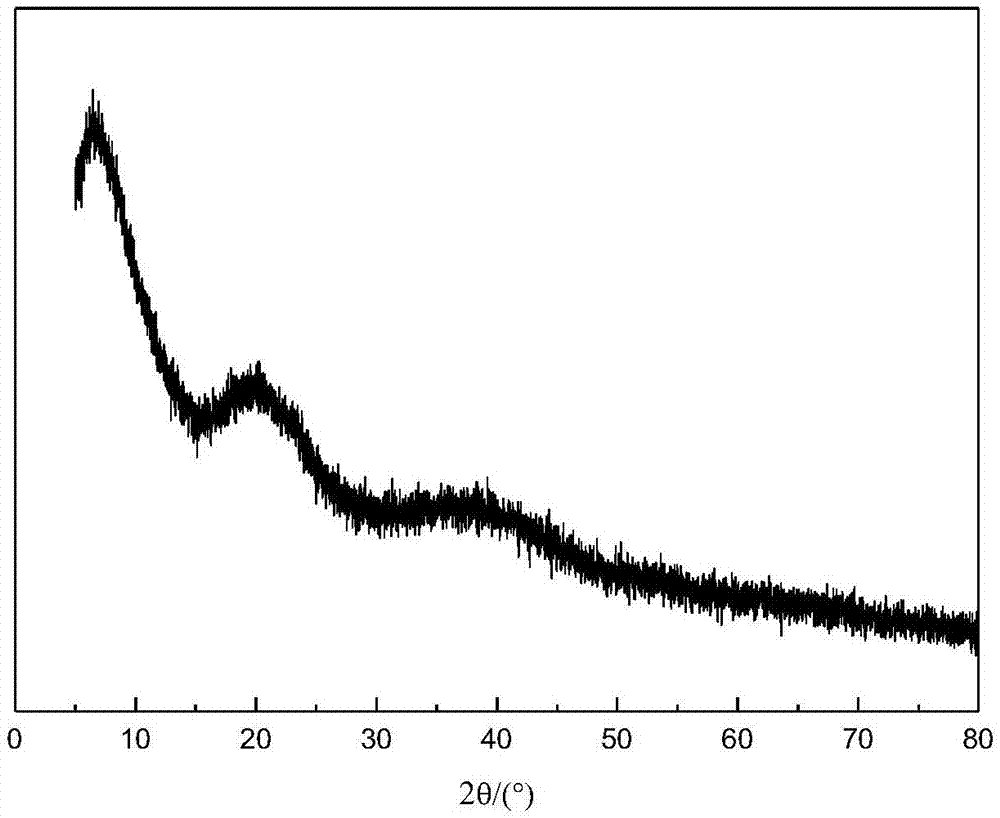
图1是制备例1所制备的固体前驱体的xrd图;
图2是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的tem图;
图3是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的xrd图;
图4是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的n2吸附-脱附等温线;
图5是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的bjh孔径分布曲线;
图6是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的xps图;
图7是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的xps中的ni2p谱图;
图8是制备例1所制备的氧掺杂碳包覆镍纳米复合材料的xps中的o1s分峰结果;
图9是制备例2所制备的氧掺杂碳包覆镍纳米复合材料的tem图;
图10是制备例2所制备的氧掺杂碳包覆镍纳米复合材料的xrd图;
图11是制备例2所制备的氧掺杂碳包覆镍纳米复合材料的bjh孔径分布曲线;
图12是制备例3所制备的固体前驱体的xrd图;
图13是制备例3所制备的氧掺杂碳包覆镍纳米复合材料的tem图;
图14是制备例3所制备的氧掺杂碳包覆镍纳米复合材料的xrd图;
图15是制备例3所制备的氧掺杂碳包覆镍纳米复合材料的bjh孔径分布曲线。
图16是制备例4所制备的固体前驱体的xrd图;
图17是制备例4所制备的严密包覆的氧掺杂碳包覆镍纳米复合材料的tem图;
图18是制备例4所制备的严密包覆的氧掺杂碳包覆镍纳米复合材料的xrd图;
图19是制备例4所制备的严密包覆的氧掺杂碳包覆镍纳米复合材料的n2吸附-脱附等温线;
图20是制备例4所制备的严密包覆的氧掺杂碳包覆镍纳米复合材料的bjh孔径分布曲线;
图21是对比例1所制备的材料的xrd图。
具体实施方式
下面根据具体实施例对本发明的技术方案做进一步说明。本发明的保护范围不限于以下实施例,列举这些实例仅出于示例性目的而不以任何方式限制本发明。
本发明的数值范围包括定义该范围的数字。短语“包含”在此用作开放端术语,基本上等效于词语“包括,但不限于”,并且短语“包含了”具有对应含义。如在此使用的,除非上下文另外明确指出,否则单数形式的“一”、“一个”以及“该”包括复数指示物。因此,例如提及“一个事物”包括多于一个这样的事物,包括基本上如在此之前所述的所有实施方案以及变体并且参考实例和附图。
在此未直接定义的任何术语应当被理解为具有与它们在本发明技术领域中通常所理解的相关联的含义。如贯穿本说明书使用的下面术语除非另外说明,应当理解为具有下面含义。
术语
术语“烷基”在本文中定义为直链或支链的烷基,例如甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基等。
术语“环烷基”在本文中定义为由单键连接并构成环状的烷基基团,例如环己基。
术语“芳基”在本文中定义为从简单芳香环衍生出的官能团或取代基,例如苯甲基。
术语“芳烃类化合物”是指分子中具有芳香环结构的一类碳氢化合物。
术语“石墨化碳层”是指在高分辨透射电镜下可明显观察到层状结构的碳结构,而非无定型结构,且层间距约为0.34nm。该石墨化碳层包覆镍纳米颗粒后形成的复合材料呈球形或类球形。
术语“介孔”定义为孔径在2~50nm范围的孔。孔径小于2nm的孔定义为微孔,大于50nm的孔定义为大孔。
术语“介孔材料”定义为包含介孔孔道结构的多孔材料。
术语“掺杂氧的石墨化碳层”中的“氧”是指氧元素,其中所述纳米复合材料的“氧含量”是指氧元素的含量,具体是指,在碳包覆纳米复合材料制备过程中,形成石墨化碳层中含有以各种形式存在的氧元素,所述“氧含量”为所有形式的氧元素的总含量。
术语“介孔分布峰”是指根据barrett-joyner-halenda(bjh)方法对脱附曲线进行计算得到的孔分布曲线上的介孔分布峰。
术语“酸洗损失率”是指制备完成的碳包覆镍的纳米复合材料产品经酸洗后镍的损失比例。其反映了石墨化碳层对镍包覆的严密程度。如果石墨化碳层对镍包覆不严密,则在酸处理后,内核的镍会被酸溶解从而流失。酸洗损失率越大,表明石墨化碳层对镍包覆的严密程度越低,酸洗损失率越小,表明石墨化碳层对镍包覆的严密程度越高。
所述的“酸洗损失率”按以下方式测量并计算:
按20ml硫酸水溶液(1mol/l)投加1g样品的比例,在90℃下对样品处理8h,然后用去离子水洗涤至中性,干燥后称重、分析,按下式计算酸洗损失率。
酸洗损失率=[1-(酸洗后复合材料中镍的质量分数×酸洗后复合材料的质量)÷(待酸洗复合材料中镍的质量分数×待酸洗复合材料的质量)]×100%。
试剂、仪器与测试
如无特殊说明,本发明所采用试剂均为分析纯,所用试剂均为市售可得。
本发明所采用xrd衍射仪的型号为xrd-6000型x射线粉末衍射仪(日本岛津),xrd测试条件为:cu靶,kα射线(波长λ=0.154nm),管电压为40kv,管电流为200ma,扫描速度为10°(2θ)/min。
本发明所采用高分辨透射电镜(hrtem)的型号为jem-2100(hrtem)(日本电子株式会社),高分辨透射电镜测试条件为:加速电压为200kv。
本发明所采用x射线光电子能谱分析仪(xps)为vgscientifc公司生产配备有avantagev5.926软件的escalab220i-xl型射线电子能谱仪,x射线光电子能谱分析测试条件为:激发源为单色化a1kαx射线,功率为330w,分析测试时基础真空为3×10-9mbar。另外,电子结合能用c1s峰(284.6ev)校正,后期分峰处理软件为xpspeak。
碳(c)、氢(h)、氧(o)三种元素的分析在elementarmicrocube元素分析仪上进行,具体操作方法和条件如下:样品在锡杯中称量1mg~2mg,放入自动进样盘,通过球阀进入燃烧管燃烧,燃烧温度为1000℃(为了去除进样时大气干扰,采用氦气吹扫),然后用还原铜对燃烧后的气体进行还原,形成二氧化碳和水。混合气体通过两根解吸柱进行分离,依次进tcd检测器检测。氧元素的分析是利用高温分解,在催化剂的作用下,将样品中的氧转化为co,然后采用tcd检测co。由于本发明的复合材料中仅含有碳、氢、氧和金属元素,因此由碳、氢、氧三种元素的总含量即可知金属元素的总含量。
不同金属元素之间的比例由x射线荧光光谱分析仪(xrf)测定,由已知的碳、氢、氧三种元素总含量,算出不同金属元素在复合材料中的含量。本发明所采用x射线荧光光谱分析仪(xrf)的型号为rigaku3013x射线荧光光谱仪,x射线荧光光谱分析测试条件为:扫描时间为100s,气氛为空气。
bet测试方法:本发明中,样品的孔结构性质由quantachromeas-6b型分析仪测定,催化剂的比表面积和孔体积由brunauer-emmett-taller(bet)方法得到,孔分布曲线根据barrett-joyner-halenda(bjh)方法对脱附曲线进行计算得到。
本发明中,金属纳米粒子的平均粒径由xrd图分峰后,由谢乐公式:d=kγ/(bcosθ)计算得到。其中k为scherrer常数,k=0.89;b为半高宽;θ为衍射角,单位弧度;γ为x射线波长,0.154054nm。
本发明提供一种环烷烃类化合物的合成方法,包括:
以碳包覆镍的纳米复合材料为催化剂,所述催化剂与芳烃类化合物在溶剂中混合后,在氢气气氛下进行加氢还原反应;化学反应方程式例示如下:
其中,所述纳米复合材料含具有壳层和内核的核壳结构,所述壳层为掺杂氧的石墨化碳层,所述内核为镍纳米颗粒,所述纳米复合材料的酸洗损失率≤60%。
在一些实施例中,所述芳烃类化合物的芳香环上还含有取代基,取代基选自c1-20的烷基、环烷基和芳基中的一种或多种。上述取代可以为单取代,也可以为多取代,包括但不限于苯、甲苯。
在一些实施例中,所述催化剂占所述芳烃类化合物质量的1-50%,优选为5-30%。
在一些实施例中,所述加氢还原反应温度一般为200-300℃。
在一些实施例中,所述氢气的压力一般为3-6mpa。
在一些实施例中,所述加氢还原反应的反应时间优选为6~10h。
在一些实施例中,所述溶剂选自醇类、醚类、烷烃类和水中的一种或多种,例如乙醇、四氢呋喃、环己烷等。
根据本发明的一个实施方式,在上述合成方法中采用的纳米复合材料为具有至少一个介孔分布峰的介孔材料。即指,该纳米复合材料在根据barrett-joyner-halenda(bjh)方法对脱附曲线进行计算得到的孔分布曲线上,至少具有一个介孔分布峰。
上述纳米复合材料,其掺杂氧的石墨化碳层表面具有丰富的缺陷位,碳材料本身就具有催化活性,与镍纳米颗粒协同发挥作用,可使该纳米复合材料具有较佳的催化性能;此外,该纳米复合材料还具有丰富的介孔结构,使其传质效率更高,从而具有更优异的催化性能。
在一些实施例中,单批次制造的复合材料,在介孔范围内有两个分布峰;如将多批次制造的复合材料混合,则在介孔范围内可以有更多的分布峰。当纳米复合材料具有不同孔径范围的多级介孔结构时,可以使其表现出更独特的性能,且多级介孔结构可适用的应用范围更广。
在一些实施例中,所述纳米复合材料的介孔结构在2~7nm的孔径范围和8~20nm的孔径范围分别具有一个介孔分布峰。
在一些实施例中,所述纳米复合材料的介孔体积占总孔体积的比例大于50%,优选大于80%。在一些实施例中,介孔体积占总孔体积的比例大于90%,甚至100%。
在一些实施例中,所述纳米复合材料的介孔体积可以为0.05-1.25cm3/g,也可以为0.30-0.50cm3/g。
在一些实施例中,所述纳米复合材料的比表面积一般大于140m2/g,可以大于200m2/g。
根据本发明的纳米复合材料,其在空气中不自燃,可以在空气中储存。
在一些实施例中,所述纳米复合材料的碳层掺杂氧元素,不掺杂氮元素。
在一些实施例中,所述纳米复合材料的碳层只掺杂氧元素,不掺杂氢、氧以外的其他元素。
在一些实施例中,所述纳米复合材料的酸洗损失率一般为≤60%,可以为≤40%,也可以为10%~20%、20%~30%或30%~40%。如上文所述,酸洗损失率反映了石墨化碳层对镍包覆的严密程度。
在一些实施例中,以质量百分比计,该纳米复合材料中,碳含量为10.0%-60.0%,镍含量为30.0%-85.0%;优选地,碳含量为15.0%-40.0%,镍含量为50.0%-80.0%。
根据本发明的纳米复合材料,在石墨化碳层中掺杂有氧。氧含量可以通过在制造过程中额外引入含氧化合物,比如多元醇来调节。通过调节所述纳米复合材料中的氧含量,可以调节石墨化碳层的催化性能。在一些实施例中,以质量百分比计,所述纳米复合材料中,氧含量小于15.0%,优选为0.2%-5.0%。
根据本发明,所述纳米复合材料中,各组分的含量之和为100%。
在一些实施例中,所述石墨化碳层的厚度为0.3nm~6.0nm,优选为0.3~3nm。
在一些实施例中,所述核壳结构的粒径为1~200nm,优选为3~100nm,更优选为4~50nm。
在一些实施例中,上述碳包覆镍的纳米复合材料通过如下方法制备:
将镍盐与多元有机羧酸及其它有机化合物在溶剂中混合形成含镍的水溶性混合物;
将所述水溶性混合物在惰性气氛或还原性气氛下高温热解。
具体地,所述的水溶性混合物是将镍盐、多元有机羧酸及可选的除前述两种外的其它有机化合物在水、乙醇等溶剂中溶解成均相溶液,然后直接蒸发除去溶剂得到含镍的水溶性混合物。本发明对蒸发溶剂的温度和工艺没有特别的限制,可以采用任意可行的现有技术,例如,在80-120℃下喷雾干燥,或在烘箱中干燥。蒸发除去溶剂的过程一般在2小时内完成,优选在1小时内完成。
在一些实施例中,根据前述的制造方法,所述的有机多元羧酸包括但不限于柠檬酸和/或对苯二甲酸;所述的镍盐包括但不限于醋酸镍;所述的其它有机化合物包括但不限于有机多元醇。
在一些实施例中,高温热解步骤中所述的惰性气氛采用氮气或氩气,所述还原性气氛可以为在惰性气氛中掺有少量氢气的气氛,其中热解过程包括升温段和恒温段,所述升温段的升温速率为0.5-10℃/min,优选2.5-10℃/min;所述恒温段的温度为400-800℃,优选500-700℃;恒温时间为20-600min,优选30-300min。
在一些实施例中,镍盐、多元有机羧酸和其它有机化合物的质量比为1:0.5-10:0-10,优选1:1-3:0-3。
又根据本发明的另一个实施方式,在上述合成方法中采用的纳米复合材料的酸洗损失率≤10%。这种严密包覆的复合材料较上述相对非严密包覆的复合材料而言,可以更好的保证内核镍在制备和应用中损失率降低,在苛刻的应用条件下保持性能的稳定性,从而更好的发挥复合材料的作用。此外,本领域通常认为催化加氢反应的活性中心是内核镍,不管催化剂的具体结构如何,必须能使反应物与镍金属中心接触。而本发明的被石墨化碳层严密包覆镍的纳米复合材料,仍具有极佳的催化加氢还原有机化合物的能力,进一步证明了石墨化碳层包裹镍内核的严密性对其催化性能十分重要,金属镍在此起到了不可或缺的修饰作用。
在该实施方式中,该“严密包覆”的碳包覆镍的纳米复合材料,可以具有介孔结构,也可以不具有介孔结构。丰富的介孔结构,更有利于催化反应的传质。在一些实施例中,该纳米复合材料具有至少一个介孔分布峰。在一些实施中,单批次制造的复合材料,在介孔范围内有两个分布峰;如将多批次制造的复合材料混合,则在介孔范围内可以有更多的分布峰。当纳米复合材料具有不同孔径范围的多级介孔结构时,可以使其表现出更独特的性能,且多级介孔结构可适用的应用范围更广。
在一些实施例中,严密包覆的所述纳米复合材料的介孔结构在2~7nm的孔径范围和8~20nm的孔径范围分别具有一个介孔分布峰。
在一些实施例中,严密包覆的所述纳米复合材料的介孔体积占总孔体积的比例大于50%,优选大于80%。在一些实施例中,介孔体积占总孔体积的比例大于90%,甚至100%。
在一些实施例中,严密包覆的所述纳米复合材料的介孔体积可以为0.05-1.25cm3/g,也可以为0.30-0.50cm3/g。
在一些实施例中,严密包覆的所述纳米复合材料的比表面积一般大于140m2/g,可以大于200m2/g。
根据本发明的严密包覆的纳米复合材料,其在空气中不自燃,可以在空气中储存。
在一些实施例中,严密包覆的所述纳米复合材料的碳层掺杂氧元素,不掺杂氮元素。
在一些实施例中,严密包覆的所述纳米复合材料的碳层只掺杂氧元素,不掺杂氢、氧以外的其他元素。
在一些实施例中,其中以质量百分比计,严密包覆的所述纳米复合材料的碳含量为15%-60%,过渡金属含量为30%-80%,优选地,碳含量为30%-60%,过渡金属含量为30%-60%。
根据本发明的纳米复合材料,在石墨化碳层中掺杂有氧。氧含量可以通过在制造过程中额外引入含氧化合物,比如多元醇来调节。通过调节所述纳米复合材料中的氧含量,可以调节石墨化碳层的催化性能,使其适用于催化不同的反应。在一些实施例中,以质量百分比计,严密包覆的该纳米复合材料中,氧含量小于15.0%,优选为1.0%-10.0%。
根据本发明,严密包覆的所述纳米复合材料中,各组分的含量之和为100%。
根据本发明的严密包覆的纳米复合材料,在一些实施例中,石墨化碳层的厚度为0.3nm~6.0nm,优选为1~3nm。在一些实施例中,所述核壳结构的粒径为1~200nm,优选为3~100nm,更优选为4~50nm。
在一些实施例中,上述严密包覆的碳包覆镍的纳米复合材料通过如下方法制备:
将镍盐与多元有机羧酸及其它有机化合物在溶剂中混合形成含镍的水溶性混合物;
将所述水溶性混合物在惰性气氛或还原性气氛下高温热解;
将经过高温热解的产物用用酸处理。
具体地,所述的水溶性混合物是将镍盐、多元有机羧酸及可选的除前述两种外的其它有机化合物在水、乙醇等中溶解成均相溶液,然后直接蒸发除去水分得到含镍的水溶性混合物。本发明对蒸发水分的温度和工艺没有特别的限制,可以采用任意可行的现有技术。例如,在80-120℃下喷雾干燥,或在烘箱中干燥。蒸发除去溶剂的过程一般在2小时内完成,优选在1小时内完成。
在一些实施例中,根据前述的制造方法,所述的有机多元羧酸包括但不限于柠檬酸和/或对苯二甲酸;所述的镍盐包括但不限于醋酸镍;所述的其它有机化合物包括但不限于有机多元醇。
在一些实施例中,高温热解步骤中所述的惰性气氛采用氮气或氩气,所述还原性气氛可以为在惰性气氛中掺有少量氢气的气氛,其中热解过程包括升温段和恒温段,所述升温段的升温速率为0.5-10℃/min,优选1-5℃/min;所述恒温段的温度为400-800℃,优选500-700℃;恒温时间为20-600min,优选30-300min。
在一些实施例中,镍盐、多元有机羧酸和其它有机化合物的质量比为1:0.5-10:0-10,优选1:1-3:0-3。
在一些实施例中,所述酸处理中优选用非氧化性强酸处理,所述非氧化性强酸包括但不限于氢氟酸、盐酸、硝酸和硫酸中的一种或其任意的组合,优选盐酸和/或硫酸。
在一些实施例中,酸处理的条件为:在30℃~100℃之间处理1h以上,优选在60℃~100℃之间处理1h~20h,更优选在70℃~90℃之间处理1h~10h。
本发明通过上述方法制备碳包覆镍纳米复合材料,而没有采用以金属-有机骨架化合物(mof)为前驱体热解的方法,该方法需要在高温、高压下于溶剂中制得具有周期性结构的晶态固体材料(即mof),通常制备mofs的条件比较严格,所需配体价格昂贵,并且很难进行大量生产;此外,该方法制备的复合材料中对金属粒子的包覆不严密,与本发明的纳米复合材料结构上有显著不同。本发明制备碳包覆镍复合材料的方法,方便在制备过程中调节石墨化碳层中的氧含量,从而方便调节纳米复合材料的电子特性,以适用于催化不同反应。
下面通过实施例来进一步说明本发明:
制备例1
(1)称取10g醋酸镍、10g柠檬酸加到含有30ml去离子水的烧杯中,在70℃下搅拌得到均相溶液,并继续加热蒸干,得到一固体前驱体。该固体的x射线衍射谱图如图1所示。
(2)将步骤(1)得到的固体至于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入流量为100ml/min的氮气,并以5℃/min的速率升温至650℃,恒温2h后停止加热,并在氮气气氛下冷却至室温,得到碳包覆镍的纳米复合材料。经x射线荧光光谱分析仪(xrf)测定,该复合材料中所含元素的质量百分含量分别为:碳24.29%,氢0.47%,氧0.96%,镍74.28%。
材料的表征:该材料的tem图如图2所示可看出,该材料为碳包覆镍的纳米复合材料,在镍纳米颗粒的外层包裹着石墨化碳层,形成完整的核壳结构。碳包覆镍的纳米材料的x射线衍射谱图如图3所示,可看出,在该材料的衍射图中存在对应于石墨碳的衍射峰(2θ角为26°)和面心立方结构(fcc)ni的衍射峰(2θ角为44.5°、51.7°和76.4°)由谢乐公式计算出该碳包覆镍纳米粒子的平均粒径为4.7nm。
bet测试表明,该复合材料的比表面积为146m2/g,孔体积为0.37cm3/g,其中>2nm的介孔体积为0.365cm3/g,占总孔体积的98.6%。图4为该复合材料的n2吸附-脱附等温线,图5为该复合材料的bjh孔径分布曲线,可以看出,所述复合材料在3.77nm和10.26nm处存在两个介孔分布峰。
该纳米复合材料的x射线光电子能谱(xps)如图6,从图6可以看到明显存在c、o、ni的xps峰,证明了o元素的有效掺杂。从图7可以看出,其中ni价态为0价。从图8可以看出,该复合纳米材料中的o不存在金属-氧(m-o)键,只存在羧基氧、羰基氧和羟基氧,充分证明了这种核壳结构有效的将高活性的ni纳米粒子与空气隔绝,核壳结构完整。
按术语部分所述方法测量、计算,本制备例制得的复合材料的酸洗损失率为36.2%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
制备例2
(1)称取10g醋酸镍,20g柠檬酸加到含有50ml去离子水的烧杯中,在80℃下搅拌得到均相溶液,并继续加热蒸干,得到一固体前驱体。
(2)将步骤(1)得到的固体置于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入流量为150ml/min的氮气,并以5℃/min的速率升温至600℃,恒温2h后停止加热,并在氮气气氛下冷却至室温,得到碳包覆镍的纳米复合材料。经元素分析仪和x射线荧光光谱分析仪(xrf)测定,该复合材料中所含元素的质量百分含量为:碳:35.64%,氢0.78%,氧3.81%,镍59.77%。
材料的表征:该材料含有以纳米金属镍为核,以石墨化碳层为壳的核壳结构,tem图如图9所示;碳包覆镍的纳米材料的x射线衍射谱图如图10所示,可看出,在该材料的衍射图中存在对应于碳的衍射峰(2θ角为26°)和fccni的衍射峰(44.5°、51.9°和76.2°),由谢乐公式计算出该碳包覆镍纳米粒子的平均粒径为34.5nm;bet测试结果见图11,表明该材料的比表面积为137m2/g,孔体积为0.343cm3/g,其中>2nm的介孔体积为0.323cm3/g,占总孔体积的94%。
按术语部分所述方法测量、计算,本制备例制得的复合材料的酸洗损失率为13.2%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
制备例3
(1)称取10g醋酸镍、10g对苯二甲酸加入30ml去离子水中,在70℃下搅拌得到均相溶液,并继续加热蒸干后得到前驱体。该固体的x衍射图如图12。
(2)将前驱体置于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入氮气,流量100ml/min,并以5℃/min的速率升温至650℃,恒温2h后停止加热,在氮气气氛下冷却至室温,得到含有碳包覆镍纳米复合材料。经元素分析仪和x射线荧光光谱分析仪(xrf)测定,该纳米复合材料中所含元素的质量百分含量为:碳29.34%;氢0.23%;氧0.56%,镍69.87%。酸洗损失率:29.4%。
材料的表征:图13是制备的纳米复合材料的tem图。可以看出,该材料含有以纳米金属镍为核,一定石墨化的碳为壳的核壳结构。在该材料的xrd衍射图中(图14)存在对应于碳的衍射峰(2θ角为25.8°)和fccni的衍射峰(44.6°、51.8°和76.4°),由谢乐公式计算出该碳包覆镍纳米粒子的平均粒径为8.4nm。说明该材料包括一定石墨化程度的碳,其中ni以面心立方结构存在。
bet测试结果表明,该纳米复合材料的比表面积为149.64m2/g,孔体积为0.29cm3/g,其中>2nm的介孔体积为0.285cm3/g,占总孔体积的98.3%。图15为该复合材料的bjh孔径分布曲线,可以看出,所述复合材料在3.87nm和18.88nm处存在两个介孔分布峰。
按术语部分所述方法测量、计算,本制备例制得的复合材料的酸洗损失率为29.4%。在术语部分所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
制备例4
(1)称取10g醋酸镍、10g柠檬酸加到含有30ml去离子水的烧杯中,在70℃下搅拌得到均相溶液,并继续加热蒸干,得到一固体前驱体。该固体的x射线衍射谱图如图16所示。
(2)将步骤(1)得到的固体置于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入流量为100ml/min的氮气,并以5℃/min的速率升温至650℃,恒温2h后停止加热,并在氮气气氛下冷却至室温,得到碳包覆镍的纳米复合材料。经元素分析仪和x射线荧光光谱分析仪(xrf)测定,该纳米复合材料中所含元素的质量百分含量分别为:碳24.29%,氢0.47%,氧0.96%,镍74.28%。
(3)按术语部分所述方法处理步骤(2)所得的纳米复合材料,干燥后得到酸洗后的纳米复合材料,即严密包覆的纳米复合材料。在所述方法的基础上,继续增加酸洗时间,酸洗损失率基本保持不变。
用元素分析仪和x射线荧光光谱分析仪(xrf)测定,该纳米复合材料中所含元素的质量百分含量为:碳44.87%,氢0.99%,氧1.81%,镍52.33%
材料的表征:该纳米复合材料的tem图如图17所示,含有以纳米金属镍为核,石墨化碳层为壳的核壳结构;碳包覆镍的纳米复合材料的x射线衍射谱图如图18所示,可看出,在该材料的衍射图中存在对应于石墨碳的衍射峰(2θ角为25.7°)和fccni的衍射峰(44.5°、51.9°和76.2°),由谢乐公式计算出该碳包覆纳米粒子的平均粒径为5.5nm;bet测试结果如图19、图20所示,该材料的比表面积为176m2/g,孔体积为0.381cm3/g,其中>2nm的介孔体积为0.376cm3/g,占总孔体积的98.7%。
对比例1:
将10g醋酸镍固体置于瓷舟内,然后将瓷舟置于管式炉的恒温区,通入流量为150ml/min的氮气,并以5℃/min的速率升温至600℃,恒温2h后停止加热,并在氮气气氛下冷却至室温,得到样品材料。经元素分析仪和x射线荧光光谱分析仪(xrf)测定,该材料中所含元素的质量百分含量为:碳1.34%,氢0.32%,氧0.18%,镍98.16%。该材料的x射线衍射谱图如图21所示,可看出,在该材料的衍射图中存在对应于fccni的衍射峰(44.2°、51.6°和76.2°)。
按术语部分所述方法测量、计算,本对比例制得的材料的酸洗损失率为100%。
实施例1
将制备例1所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.1g复合材料、1.85g反应物甲苯,100ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为6mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间10小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过以下公式计算反应物转化率及目的产物选择性:
转化率=已反应的反应物质量/反应物加入量×100%
选择性=目的产物质量/反应生成物质量×100%
经分析后,得甲苯转化率为98.3%,甲基环己烷选择性为97.9%。
实施例2
将制备例1所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.1g复合材料、0.332g反应物甲苯,30ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为98.2%,甲基环己烷选择性为99.5%。
实施例3
将制备例1所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.1g复合材料、0.498g反应物甲苯,50ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度300℃,反应预定时间6小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为95.2%,甲基环己烷选择性为99.1%。
实施例4
将制备例1所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.1g复合材料、0.645g甲苯,50ml水加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为4mpa,搅拌升温,升温至预定反应温度300℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为94.6%,甲基环己烷选择性为99.1%。
实施例5
将制备例1所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.1g复合材料、0.39g苯,30ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得苯转化率为95.4%,环己烷选择性为99.2%。
实施例6
将制备例2所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.2g复合材料、0.664g甲苯,50ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为96.1%,甲基环己烷选择性为98.9%。
实施例7
将制备例3所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.2g复合材料、0.664g甲苯,50ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为97.4%,甲基环己烷选择性为99.4%。
实施例8
将制备例4所得复合材料作为催化剂用于芳烃类化合物反应物加氢制备目的产物环烷烃类化合物的反应,具体的实验步骤为:
将0.2g复合材料、0.664g甲苯,50ml环己烷加入反应釜中,通h2置换反应釜3次后,通h2使反应釜内压力为3mpa,搅拌升温,升温至预定反应温度200℃,反应预定时间8小时后停止加热,降至室温,排压,开反应釜取产物甲基环己烷进行色谱分析。通过实施例1所列公式计算反应物转化率及目的产物选择性:
经分析后,得甲苯转化率为95.3%,甲基环己烷选择性为99.6%。
可见,采用本发明的碳包覆镍纳米复合材料催化芳烃类化合物加氢合成环烷烃类化合物,在宽的温度范围和压力范围内,均可实现反应转化率94%,且环烷烃类化合物的选择性均可达到97%以上,材料的催化性能稳定,表现了良好的重复性、高活性及高选择性。
本领域技术人员应当注意的是,本发明所描述的实施方式仅仅是示范性的,可在本发明的范围内作出各种其他替换、改变和改进。因而,本发明不限于上述实施方式,而仅由权利要求限定。
- 还没有人留言评论。精彩留言会获得点赞!