盐酸奥达特罗晶型B及其制备方法与流程
本发明属于医药领域,具体涉及一种盐酸奥达特罗新的晶型及其制备方法。
背景技术:
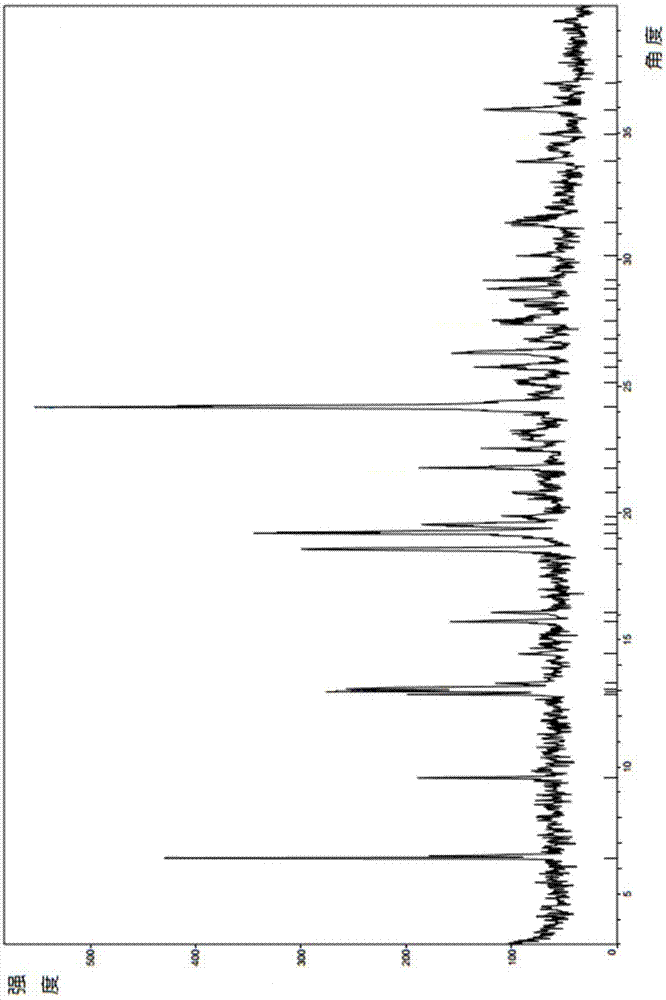
盐酸奥达特罗,系统命名为:6-羟基-8-{(1r)-1-羟基-2-{[2-(4-甲氧基苯基)-1,1-二甲基乙基]氨基}乙基}-4h-苯并[1,4]噁嗪-3-酮盐酸盐(6-hydroxy-8-{(1r)-1-hydroxy-2-{[2-(4-methoxyphenyl)-1,1-dimethyl-ethyl]amino}ethyl}-4h-[1,4]-benzoxazin-3-one,,hydrochloride),分子式:c21h27cln2o5,分子量:386.44,cas登记号:868049-49-4,结构式如1所示盐酸奥达特罗是一种长效β2受体激动剂,适用于慢性阻塞性肺病(copd)患者。公开号cn101817800a(公开日2010年9月1日)的中国发明专利申请说明书中记载了未区分对映体的6-羟基-8-{1-羟基-2-{[2-(4-甲氧基苯基)-1,1-二甲基乙基]氨基}乙基}-4h-苯并[1,4]噁嗪-3-酮及其制备方法和用途。公开号cn102827097a(公开日2012年12月19日)的中国发明专利申请说明书披露了结构式1所示的纯对映异构体,同时披露了如下的制备方法:“将5.25克(17.7毫摩尔)6-苄氧基-8-(r)-环氧乙烷基-4h-苯并[1,4]噁嗪-3-酮和6.30克(35.1毫摩尔)2-(4-甲氧基-苯基)-1,1-二甲基-乙基胺与21毫升异丙醇混合,并在密闭反应容器内在微波辐射下于135℃搅拌30分钟。将溶剂蒸发掉,并将残余物进行色谱处理(氧化铝;乙酸乙酯/甲醇梯度)。将如此得到的产物进一步通过由二乙醚/二异丙醚混合物再结晶纯化”,得到6-苄氧基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮。然后“将5.33克(11.2毫摩尔)6-苄氧基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮在120毫升甲醇中的悬浮液与0.8克钯/炭(10%)混合,加热至50℃,在3巴氢气压下氢化。然后抽滤催化剂,并蒸干滤液。将残余物溶于20毫升异丙醇中,并加入2.5毫升溶于异丙醇的5摩尔盐酸。该产物用200毫升二乙醚沉淀,抽滤并干燥,”即得到6-羟基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮-盐酸盐(盐酸奥达特罗)。但是发明人发现按照上述方法,盐酸奥达特罗在抽滤时极易吸潮转变为油状物,无法得到稳定的结晶产品,给后续的质控和制剂工作造成了很大的困难。众所周知,同一种药物,不同晶型之间溶解度、稳定性、流动性、可压缩性、生物利用度等各方面的理化性质都可能存在很大差异,从而影响药物的疗效。因此,需要开发具有优越的生理化学特性的盐酸奥达特罗的新晶型,从而有利于制剂加工和临床应用。技术实现要素:为了克服现有技术的不足,本发明提供一种新的盐酸奥达特罗的晶型,盐酸奥达特罗的晶型b。该晶型不易吸潮,稳定性显著提高,便于产品的质量控制;而且制备工艺简洁,有利于工业化生产中的成本控制。为了实现上述发明目的,本发明采用了如下的技术方案:一种结构式1所示的盐酸奥达特罗的晶型b,其中使用cu-kα辐射,以2θ角度表示的x射线衍射图谱在6.40°±0.2°、9.57°±0.2°、12.85°±0.2°、12.97°±0.2°、13.07°±0.2°、18.58°±0.2°、19.21°±0.2°、19.54°±0.2°、21.79°±0.2°、24.18°±0.2°、26.31°±0.2°和35.90°±0.2°处有特征吸收峰。优选的,所述盐酸奥达特罗的晶型b使用cu-kα辐射,以2θ角度表示的x射线衍射图谱在6.40°±0.2°、9.57°±0.2°、12.85°±0.2°、12.97°±0.2°、13.07°±0.2°、13.29°±0.2°、18.58°±0.2°、19.21°±0.2°、19.54°±0.2°、19.88°±0.2°、20.79°±0.2°、21.79°±0.2°、24.18°±0.2°、25.76°±0.2°、26.31°±0.2°、27.59°±0.2°、28.40°±0.2°、28.84°±0.2°、29.17°±0.2°、30.14°±0.2°、31.44°±0.2°、33.86°±0.2°和35.90°±0.2°处有特征吸收峰。优选的,所述盐酸奥达特罗的晶型b使用cu-kα辐射,以2θ角度表示的x射线衍射图谱与图1基本一致。本发明还有一个目的在于提供上述盐酸奥达特罗的晶型b的制备方法,包括如下步骤:i.将盐酸奥达特罗粗品和有机溶剂投入反应容器中,搅拌,升温,使溶清,保温继续搅拌;ii.降低温度,加入晶种,搅拌,析晶,过滤,干燥,即得。优选的,所述的有机溶剂选自甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、dmf、nmp、dmso、甲苯、正丙醇中的一种或一种以上。更优选的,所述有机溶剂为异丙醇或异丙醇-甲醇;其中异丙醇和甲醇的体积比为:异丙醇:甲醇=7:1~15:1;更优选的,异丙醇:甲醇=10:1。优选的,所述盐酸奥达特罗粗品和所述有机溶剂的重量体积比为:盐酸奥达特罗粗品:所述有机溶剂=1g:8ml~1g:15ml;更优选的,所述盐酸奥达特罗粗品和所述有机溶剂的重量体积比为:盐酸奥达特罗粗品:所述有机溶剂=1g:11ml。优选的,所述步骤i中,升温至50~55℃,溶清后保温搅拌20min~1h,优选0.5h。优选的,所述步骤ii中,降温至25~30℃,加入晶种后在25~30℃下搅拌1h~3h,优选2h。上述制备方法中,所述晶种通过如下步骤制备:i.将盐酸奥达特罗粗品和异丙醇投入反应容器中,搅拌,升温至50-55℃,使溶清,保温继续搅拌半小时;其中所述盐酸奥达特罗和异丙醇的重量体积比为:盐酸奥达特罗粗品:异丙醇=1g:8ml~1g:15ml;ii.降低温度至25~30℃,保温30min~1h,优选0.5h;iii.再次升温至50~55℃,保温搅拌0.5h~3h,优选1h;iv.再次降温至25~30℃,保温搅拌1h~3h,优选2h,过滤,干燥,即得。上述盐酸奥达特罗粗品可以按照下面的实施例1的步骤1~3所述的方法制备得到。本发明第三个目的在于提供一种药物组合物,包括上述的盐酸奥达特罗的晶型b或上述的制备方法制备得到的盐酸奥达特罗的晶型b,和药学上可以接受的辅料。优选的,所述药物组合物为吸入气雾剂、吸入粉雾剂或吸入溶液。此外,本发明还有一个目的在于提供上述盐酸奥达特罗的晶型b、上述制备方法制备得到的盐酸奥达特罗的晶型b、或上述药物组合物在制备治疗copd药物中的用途。本发明所述盐酸奥达特罗晶型b不易吸潮,稳定性较现有技术显著提高,从而有利于产品的质量控制以及后续的制剂加工。此外,本发明制备的奥达特罗晶型b在水中的溶解度与原研晶型相比显著提高,可改善其生物利用度从而大幅度提高药效。同时,本发明提供的所述晶型的制备工艺简洁,有利于工业化生产中的成本控制,具有较高的经济价值。附图说明下面结合附图,对本发明作进一步的说明。图1为实施例1制备的盐酸奥达特罗晶型b的x衍射图谱。具体实施方式以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。此外应理解,在阅读本文讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。其中,部分试剂、原料、仪器购买情况如下:6-苄氧基-8-((r)-2-氯-1-羟基-乙基-4h-苯并[1,4]-噁嗪-3-酮,cas869478-11-5,纯度98%和2-(4-甲氧基-苯基)-1,1-二甲基-乙基胺盐酸盐cas56490-93-8,纯度99%,均采购自上海有吉医药科技有限公司。核磁共振仪:brukeravanceiiihd500;质谱仪:ltqorbitrapelite;液相色谱仪:agilent1260。在下述实施例及对比例中,hplc检测均按照如下方法进行:色谱柱:watersxbridge5μm(4.6*250mm);流动相:a:0.1%庚烷磺酸钠(ph3.2)水溶液和b:乙腈,梯度洗脱程序如表1所示:表1hplc梯度洗脱程序time(min)a%b%073273692723495143356555326855.17327607327流速:1.0ml/min;柱温:30℃;检测波长:220nm;进样体积:10μl;稀释液:乙腈。在下述实施例和对比例中,x射线衍射的检测条件为:仪器型号:brukerd8advance测试条件:x-射线源:cu-k工作电流:40ma工作电压:40kv检测器:psd初始角度:4°(2-θ)结束角度:40°(2-θ)增量:0.05°/step扫描速度:1sec/step实施例1盐酸奥达特罗晶型b的制备1)6-苄氧基-8-(r)-环氧乙烷基-4h-苯并[1,4]噁嗪-3-酮的制备将100.6g(0.3mol)6-苄氧基-8-((r)-2-氯-1-羟基-乙基-4h-苯并[1,4]-噁嗪-3-酮和2ldmf加入至反应瓶中,搅拌,冰浴降温,降至0℃,加入400ml2n氢氧化钠水溶液,0~5℃保温反应四小时,将该反应液倒入至冰水中,0~5℃搅拌1小时,过滤,滤饼50℃真空干燥得86g类白色固体控。收率96%,纯度hplc:96.5%.2)6-苄氧基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮的制备将步骤1)制备得到的52.5g(0.178mol)6-苄氧基-8-(r)-环氧乙烷基-4h-苯并[1,4]噁嗪-3-酮,63g(0.351mol)2-(4-甲氧基-苯基)-1,1-二甲基-乙基胺和500ml异丙醇加入至反应瓶中,回流反应24小时,降至20~30℃,然后加入30g(0.3mol)浓盐酸,0~5℃搅拌1小时,过滤,滤饼用500ml无水甲醇打浆得60g类白色固体,收率:66.2%,纯度95.8%3)盐酸奥达特罗粗品的制备将步骤2)制备得到的60g(0.117mol)6-苄氧基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮,600ml甲醇和8g10%钯碳加入至反应瓶中,加热至50℃,在3巴氢气压下氢化。然后抽滤除去催化剂,滤液50℃真空蒸干溶剂得49.1g白色泡状固体,收率:99.27%,纯度:95.3%。4)盐酸奥达特罗晶种的制备将步骤3)制备得到的1g盐酸奥达特罗粗品和9ml异丙醇加入至反应瓶中,搅拌,升温至50~55℃,使溶清,保温搅拌半小时;降温至30℃,有固体析出,得到混悬液,保温半小时;然后再升温至50~55℃,保温搅拌1小时,然后再降温至25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得0.8g类白色固体,收率:80%,纯度:99.8%。晶体x射线粉末衍射数据与表2所示一致。5)盐酸奥达特罗晶型b的制备将5g步骤2)制备得到的盐酸奥达特罗粗品和45ml异丙醇加入至反应瓶中,搅拌,升温至50~55℃,溶清,保温搅拌半小时,降温至30℃,加入晶种,25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得4.5g类白色固体,收率:90%,纯度:99.8%。1hnmr(dmso-d6):δppm1.19(d,6h),2.90(m,3h),3.14(m,1h),3.74(s,3h),4.60(m,2h),5.15(m,1h),6.10(d,1h),6.37(d,1h),6.58(d,1h),6.90(d,2h),7.15(d,2h),8.58(m,1h),8.89(m,1h),9.33(s,1h),10.65(s,1h).ms(esi)387m/z(m-hcl+h)+。本实施例得到的盐酸奥达特罗晶型b的x射线衍射图谱如图1所示,具体结果见表2。表2盐酸奥达特罗晶型b的x射线粉末衍射数据按照《中国药典》(2005版)二部附录xixj药物引湿性试验指导原则进行试验,测定本实施例制备得到的盐酸奥达特罗晶型b的引湿性,结果引湿性增重0.18%。室温下测定,晶体b在水中的溶解度为1000mg/10ml。实施例2盐酸奥达特罗晶型b的制备将5g实施例1得到的盐酸奥达特罗粗品,50ml异丙醇和5ml甲醇加入至反应瓶中,搅拌,升温至50~55℃,使溶清,保温搅拌半小时,降温至30℃,加入实施例1制备的晶种,25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得4.6g类白色固体,收率:92%,纯度:99.7%。采用brukerd8advancex射线衍射仪,本实施例的产物得到与图1基本一致的x射线衍射图谱(图谱略)。按照《中国药典》(2005版)二部附录xixj药物引湿性试验指导原则进行试验,测定本实施例制备得到的盐酸奥达特罗晶型b的引湿性,结果引湿性增重0.33%。室温下测定,晶体b在水中的溶解度为1000mg/10ml。实施例3盐酸奥达特罗晶型b的制备将4.3g实施例1得到的盐酸奥达特罗粗品,60ml异丙醇和4ml甲醇加入至反应瓶中,搅拌,升温至50~55℃,使溶清,保温搅拌半小时,降温至30℃,加入晶种,25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得3.8g类白色固体,收率:88%,纯度:99.3%。采用brukerd8advancex射线衍射仪,本实施例的产物得到与图1基本一致的x射线衍射图谱(图谱略)。按照《中国药典》(2005版)二部附录xixj药物引湿性试验指导原则进行试验,测定本实施例制备得到的盐酸奥达特罗晶型b的引湿性,结果引湿性增重0.25%。室温下测定,晶体b在水中的溶解度为1000mg/10ml。实施例4盐酸奥达特罗晶型b的制备将8g实施例1得到的盐酸奥达特罗粗品,56ml异丙醇和8ml甲醇加入至反应瓶中,搅拌,升温至50~55℃,使溶清,保温搅拌半小时,降温至30℃,加入晶种,25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得7g类白色固体,收率:87%,纯度:99.4%。采用brukerd8advancex射线衍射仪,本实施例的产物得到与图1基本一致的x射线衍射图谱(图谱略)。按照《中国药典》(2005版)二部附录xixj药物引湿性试验指导原则进行试验,测定本实施例制备得到的盐酸奥达特罗晶型b的引湿性,结果引湿性增重0.33%。室温下测定,晶体b在水中的溶解度为1000mg/10ml。实施例5盐酸奥达特罗晶型b的制备将5g实施例1得到的盐酸奥达特罗粗品,55ml乙酸乙酯加入至反应瓶中,搅拌,升温至50~55℃,使溶清,保温搅拌半小时,降温至30℃,加入晶种,25~30℃保温搅拌2小时,过滤,滤饼50℃真空干燥得4.5g类白色固体,收率:90%,纯度:99.4%。采用brukerd8advancex射线衍射仪,本实施例的产物得到与图1基本一致的x射线衍射图谱(图谱略)。按照《中国药典》(2005版)二部附录xixj药物引湿性试验指导原则进行试验,测定本实施例制备得到的盐酸奥达特罗晶型b的引湿性,结果引湿性增重0.22%。室温下测定,晶体b在水中的溶解度为1000mg/10ml。对比例1盐酸奥达特罗的制备按照公开号cn102827097的中国发明专利申请记载的方法(实施例1)制备盐酸奥达特罗,步骤如下所示:将5.33克(11.2毫摩尔)6-苄氧基-8-{(r)-1-羟基-2-[2-(4-甲氧基-苯基)-1,1-二甲基-乙基氨基]-乙基}-4h-苯并[1,4]噁嗪-3-酮在120毫升甲醇中的悬浮液与0.8克钯/炭(10%)混合,加热至50℃,在3巴氢气压下氢化。然后抽滤催化剂,并蒸干滤液。将残余物溶于20毫升异丙醇中,并加入2.5毫升溶于异丙醇的5摩尔盐酸,该产物用200毫升二乙醚沉淀,抽滤并干燥。但是在抽滤时,发现盐酸奥达特罗极易吸潮转变为油状物,无法得到稳定的结晶产品。对比例2盐酸奥达特罗的制备按照公开号cn101208316a的中国发明专利申请实施例1记载的方法制备盐酸奥达特罗。产率63-70%,纯度:98.5%。制备得到的盐酸奥达特罗的晶型使用cu-kα辐射,以2θ角度表示的x射线衍射图谱在11.91°±0.2°、15.19°±0.2°、16.88°±0.2°、17.34°±0.2°、19.04°±0.2°、19.71°±0.2°、21.02°±0.2°、21.35°±0.2°、22.15°±0.2°、23.21°±0.2°、28.41°±0.2°、29.79°±0.2°和30.71°±0.2°处有特征吸收峰。该晶体室温下在水中的溶解度为900mg/10ml。按照现有技术制备得到的盐酸奥达特罗,晶型与本发明的不同,在水中的溶解度也不及本发明的晶型b。总之,本发明提供了一种盐酸奥达特罗的新晶型,该晶型不易吸潮,稳定性和在水中的溶解度显著提高,便于产品的质量控制、后续的制剂加工,同时可提高药效。同时,本发明提供的所述晶型的制备工艺简洁,有利于工业化生产中的成本控制,具有较高的经济价值。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1