一种噁嗪酮类化合物及其合成方法与流程
本发明涉及化工领域,具体涉及一种噁嗪酮类化合物及其合成方法。
背景技术
癌症已经成为影响人类健康与寿命的主要原因之一,目前癌症的主要治疗方式是放、化疗,但放、化疗具有一定的毒副作用,在杀死癌细胞的同时也对正常细胞产生很大的伤害,甚至正常细胞也会被杀死,严重影响了患者的生存质量,以至威胁生命,文献报道,噁嗪酮类化合物具有抗病毒、抗肿瘤,抗真菌和驱虫等作用噁嗪酮类化合物是禾本科植物的重要次生代谢产物,是非常重要的天然产物,具有抗菌谱广和较低生物毒性等优点,但是从植物中获得的量很少,且结构单一,为了更好的保护自然资源,科学家们对噁嗪酮类化合物进行了大量的研究,以期能筛选高效低毒的噁嗪酮类化合物。过去几十年里,研究者对这类化合物的合成路线进行了大量的研究,主要有以下几种方法:
1、邻氨基苯酚与邻二卤代物间的亲核取代反应,由于邻二卤代物是一种毒性非常强的物质,对生产过程中车间工人的危害极大。
2、邻卤代苯酚与缩合剂间的环合反应,这类方法多采用重金属作催化剂,成本高、反应时间较长。
3、邻硝基苯酚经过硝基还原后与环合试剂之间的环合反应,这类方法大多步骤繁多,中间体不太稳定,杂质多,总收率普遍较低。
技术实现要素:
本发明针对现有噁嗪酮类化合物及其合成方法的不足,提供一种原料简单,操作安全,产物纯度高的噁嗪酮类化合物及其合成方法。
一种噁嗪酮类化合物,其化学结构式ⅱ为:
其中,r1为芳基、取代芳基中的一种,r2为氢、c1-6烷基、c1-6烷氧基、羟基中的一种。
进一步,步骤二中所述的c1-6烷基为甲基、乙基、正丁基、异丁基中的一种。通过该方案,可以得到更多不同结构的产物,他们的生物相容性或理化性质会稍有区别,可以从中筛选高效低毒的目标化合物。
进一步,步骤二中所述c1-6烷氧基为甲氧基、乙氧基中的一种。通过该方案,可以进一步得到更多不同结构的产物,他们的生物相容性或理化性质会稍有区别,可以从中筛选高效低毒的目标化合物。
进一步,所述噁嗪酮类化合物用于制备抗卵巢癌药物的用途。
本方案中的噁嗪酮类化合物的合成方法,包括以下步骤:
步骤一、向反应器中加入0.08~0.12mol的双甲酮、0.8~1.2mol的乙腈、0.08~0.12mol的对甲苯磺酰叠氮及0.08~0.12mol的碱金属催化剂,在常温条件下反应5h,过滤,滤液减压浓缩至干后用柱色谱法分离纯化;再用体积比为1:4的乙酸乙酯/石油醚重结晶即得中间体i:
5,5-二甲基-2-重氮-1,3-环己二酮;
步骤二、向反应器中加入醛、胺反应0.5h,再加入上述中间体ⅰ、反应0.5h,采用体积比为2:3的乙酸乙酯/水溶液将反应液萃取,取乙酸乙酯层减压浓缩,再用tlc分离,得到目标化合物式ⅱ。
本发明和现有技术相比较具有如下优点和效果:
本发明提供了一种全新的合成路线,克服了现有噁嗪酮类化合物及其合成方法的不足,在常温常压下即可完成,工艺简单,操作安全,产物结构多样,产物纯度高,结构丰富、生物相容性好。
本发明的方法中,反应条件简单,中间体相对比较稳定,工艺安全高效,收率较高。
进一步,步骤一中所述的碱性催化剂为koh或kco3。优选这两种碱性催化剂都比较廉价、易得。
进一步,步骤二所述反应器为微波反应器,微波反应器的功率100-140w。该优化方案具有能够节约反应时间,提高产率和效率的有益效果。
附图说明
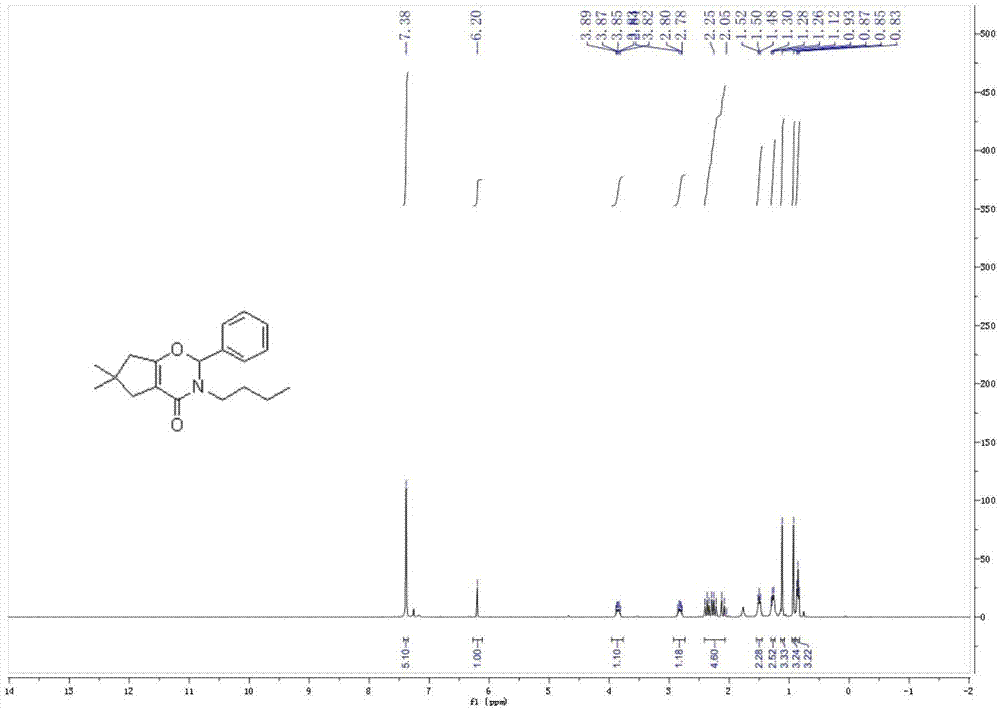
图1为本发明噁嗪酮类化合物及其合成方法实施例1的核磁共振氢谱(1hnmr);
图2为本发明噁嗪酮类化合物及其合成方法实施例1的核磁共振碳谱(13cnmr);
图3为本发明噁嗪酮类化合物及其合成方法实施例2的核磁共振氢谱(1hnmr);
图4为本发明噁嗪酮类化合物及其合成方法实施例2的核磁共振碳谱(13cnmr);
图5为本发明噁嗪酮类化合物及其合成方法实施例3的核磁共振氢谱(1hnmr);
图6为本发明噁嗪酮类化合物及其合成方法实施例3的核磁共振碳谱(13cnmr)。
具体实施方式
下面通过具体的实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,并非将本发明的保护范围局限于此。
实施例1
3-丁基-6.6二甲基-2-苯基-2,3,4,6-四氢环戊烷并[1.3]噁嗪-4(5h)酮的制备,合成路线为:
步骤为:
1)合成中间体5,5-二甲基-2-重氮-1,3-环己二酮
在500ml圆底烧瓶中,分别加入双甲酮(14g,0.1mol)、乙腈(60ml,1mol)、对甲苯磺酰叠氮(tsn3)(19.8g,0.1mol)和无水碳酸钾(11g,0.1mol),在常温条件下反应5h,将反应物过滤,用适量ch2cl2润洗,滤液浓缩后加入乙酸乙酯/石油醚(1:4)20ml、活性炭0.5g,振荡脱色,抽滤,浓缩,采用柱色谱法分离纯化,重结晶,得到淡黄色晶体。产率92%.13cnmr(100mhz、cdcl3)28.35,31.12,50.52~77.35,189.89;
2)3-丁基-6.6-二甲基-2-苯基-2.3.6.7四氢环戊烷并[1.3]噁嗪-4(5h)-酮
在25ml三颈圆底烧瓶中,先加苯甲醛(0.0061mol)、正丁胺(0.0061mol)、适量乙醇反应0.5h,,再加入5,5-二甲基-2-重氮-1,3-环己二酮(0.0061mol)、甲苯作溶剂,在氩气环境中,130℃微波催化反应0.5h。将反应液萃取(乙酸乙酯/水=2/3),取乙酸乙酯层减压浓缩,再用硅胶薄层板分离得到淡黄色油状液体。
本方法所获噁嗪酮类化合物的收率58%。
核磁共振波谱数据为:
1hnmr(400mhz,cdcl3)δ7.38(s,5h),6.20(s,1h),3.85(dt,j=15.0,7.6hz,1h),2.92–2.73(m,1h),2.26(m,4h),1.54–1.45(m,2h),1.31–1.24(m,2h),1.12(s,3h),0.93(s,3h),0.85(t,j=7.3hz,3h).
13cnmr(101mhz,cdcl3)δ165.08,163.08,136.73,129.35,128.45,127.09,109.49,89.89,77.33,77.01,76.69,46.12,43.49,40.96,36.37,30.68,29.80,20.03,13.73.
实施例2
3-异丁基-6.6-二甲基-2-苯基-2.3.6.7四氢环戊烷并[1.3]噁嗪-4(5h)-酮的制备。
在25ml圆底烧瓶中,先加苯甲醛(0.0061mol)、异丁胺(0.0061mol)、适量乙醇反应0.5h,再加入5,5-二甲基-2-重氮-1,3-环己二酮(0.0061mol)、甲苯,在氩气环境中,130℃微波催化反应0.5h,将反应液萃取(乙酸乙酯/水=2/3),取乙酸乙酯层减压浓缩,再用硅胶薄层板分离得到淡黄色油状液体。
本方法所获噁嗪酮类化合物的收率49%。
核磁共振波谱数据为:
1hnmr(400mhz,cdcl3)δ7.32(s,5h),6.18(s,1h),3.98–3.84(m,1h),2.53–2.44(m,1h),2.36(dd,j=14.5,1.3hz,1h),2.34(dd,j=17.1hz,1h),2.15(dd,j=14.5,1.3hz,1h),2.03(d,j=17.1hz,1h),1.94–1.83(m,1h),1.08(s,3h),0.94–0.87(m,6h),0.86–0.84(m,3h).
13cnmr(101mhz,cdcl3)δ164.84,163.08,136.94,129.15,128.38,126.82,109.52,89.82,51.14,46.12,40.89,36.42,29.71,27.89,20.10.
实施例3
3-正己基-6.6-二甲基-2-苯基-2.3.6.7四氢环戊烷并[1.3]噁嗪-4(5h)-酮的制备。
在25ml三颈圆底烧瓶中,加苯甲醛(0.0061mol)、正己胺(0.0061mol)、乙醇等反应0.5h,再加入5,5-二甲基-2-重氮-1,3-环己二酮(1.0126g,0.0061mol)、甲苯,在氩气环境中,130℃微波催化反应0.5h,将反应液萃取(乙酸乙酯/水=2/3),取乙酸乙酯层减压浓缩,再用硅胶薄层板分离(展开剂:乙酸乙酯/石油醚=1/4),得到淡黄色油状液体。
本方法所获噁嗪酮类化合物的收率37%。
核磁共振波谱数据为:
1hnmr(400mhz,cdcl3)δ7.38(s,5h),6.20(s,1h),3.88–3.78(m,1h),2.86–2.76(m,1h),2.40–2.08(m,6h),1.53–1.47(m,2h),1.25–1.20(m,6h),1.11(s,3h),0.93(s,3h),0.83(dd,j=7.0,4.9hz,3h).
13cnmr(101mhz,cdcl3)δ165.14,163.08,136.69,129.36,128.46,127.09,109.46,89.89,46.10,43.78,40.96,36.38,31.40,29.81,28.49,26.45,22.47,14.00.以上实施例中用到的各种试剂的摩尔数均在0.8~1.2倍范围内有效。
生物活性测试:
将卵巢瘤细胞(skov-3细胞)以3×103个/孔接种于96孔细胞培养板,待细胞贴壁后(约24h),设置y-1及y-2两个实验组,每组的供试品浓度均为0.05,0.1,0.5,1,5,10,50,100,200ug/ml,每个实验组均对应一个空白对照组,其中y-1组使用噁嗪酮类化合物i作为抑制剂,y-2组采用噁嗪酮类化合物ⅱ作为抑制剂。继续孵育48h后,每孔加入20μl浓度为5mg·ml-1的3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐溶液(mtt)。振荡后继续孵育4h,弃去上清液,加入二甲基亚砜(dmso)200μl,振摇,酶标仪测定a570(570nm处的吸收光度),计算细胞生长抑制率和ic50,统计结果见表1及表2。
表1:
表2:
通过上述表1及表2的数据可以看出,本发明中的噁嗪酮类化合物1,在细胞浓度为200ug/ml时,其对卵巢癌细胞的抑制率能够达到66.57%以上,通过计算其ic50为197ug/ml,利用本发明中的噁嗪酮类化合物2,在细胞浓度为200ug/ml时,其对卵巢癌细胞的抑制率能够达到64.57%以上,通过计算其ic50为146ug/ml,说明该类化合物对卵巢癌细胞具有较强的抑制作用。
- 还没有人留言评论。精彩留言会获得点赞!