利用CRISPR/Cas9技术创制不包颈水稻两系不育系的方法与流程
本发明涉及植物转基因技术领域和作物遗传育种领域,具体地说,涉及一种利用crispr/cas9技术对水稻eui基因定点突变的方法。
背景技术:
杂交水稻的研究与应用为我国粮食安全、农民增收和种业发展做出了巨大的贡献。不断运用新技术、新方法将已克隆的功能基因转化为育种家所能利用的分子设计育种,以提高杂种优势、降低种子生产成本是杂交水稻研究的重要方向。目前生产上应用的水稻不育系抽穗时普遍存在不同程度的包颈现象,极大地制约不育系异交结实率和繁殖制种产量。虽然生产上可应用喷施赤霉素的方法能克服不育系的包穗现象,但赤霉素的使用提高种子生产成本,降低种子质量,且容易造成环境污染。因此,实现遗传解除不育系包穗障碍一直为育种家所追求的。
20世纪80年代初,由美国学者rutger等发现并命名的高秆隐性突变体eui(elongateduppermostinternode)基因为生产上解决不育系包颈遗传障碍创造了有利的条件。eui是编码细胞色素氧化物类酶合成的一个基因,催化ga的分解代谢,在抽穗期的幼穗和最上节间表达强烈。eui基因突变可导致编码合成的酶失活,使得突变体幼穗和最上节间中积累大量的ga,从而引起最上节间的剧烈伸长。由于这种上部节间显著伸长的特性可克服杂交水稻不育系稻穗不能完全伸出叶鞘的包穗障碍,同时也可使恢复系株高和穗颈节增高,有利于提高杂交水稻制种产量,故具有十分重要的育种价值。
自eui基因发现以来,水稻育种学家一直致力于将该基因应用于杂交水稻,用以解除不育系包颈障碍,减少制种时使用赤霉素所造成的环境污染,降低种子成本,提高制种产量。但由于技术条件的限制,在探索eui基因的育种应用过程中主要使用杂交、回交法和直接诱变法两种常规的育种方法。杂交、回交法是通过杂交和多次回交将高秆隐性突变体eui基因转移到优良杂交水稻不育系中,从而选育出吐颈的杂交稻不育系。如申宗坦等(1987)和何祖华等(1991)用籼型三系不育系珍籼97a转育的吐珍长a,王才林用两系光温敏培矮64s培育出的p8hs。直接诱变法是用γ-射线辐射或化学诱变剂处理生产上应用的三系杂交水稻保持系、两系杂交水稻不育系,从诱变后代中选育具有目标性状的吐颈不育系。如杨仁崔等用金23b辐射后经回交育成的金23ea,周广洽等用培矮64s辐射育成的双低培es。虽然这两种方法都能从遗传上克服不育系的包颈问题,但均存在很大的弊端。杂交、回交法由于难以完全保留原不育系综合的优良性状,且存在育种难度大、周期长、效率低弊端,至今尚无可接近生产应用的选育成果报道;而直接诱变法产生的变异缺乏定向性,且有益突变的频率极低(10万分之1.5),只有大规模种植诱变后代,才有选择到目标性状的机会,而大规模种植诱变群体必然会导致花费更多的人力和物力,给育种工作带来许多不便之处。因此,发明一种高效、快速、精确定向改良不育系包颈遗传障碍,且又能保持原不育系优良性状新技术,对于促进杂交水稻的发展具有重要意义。
crispr-cas9基因编辑技术是近年来发展和快速应用的定点基因编辑技术,具有构建简单,使用方便,成本低廉,能够实现全基因组编辑和多靶点编辑,特异性和敲除效率非常高,遗传编辑之后不留下外源基因污染等一系列优势,现已成为应用最广泛的基因组编辑技术,在作物遗传改良和品种培育上具有重大应用潜力。国内外学者利用crispr/cas9技术在改良水稻产量、品质、抗性、育性、株型等重要性状已取得很大的进展,如直立密穗osdep1、大穗osckx2、穗粒数gnla、粒重gw、粒长gs3、香味osbadh2、糯性oswaxy、稻瘟病oserf922、光敏核雄性不育csa、温敏核雄性不育tms5、半矮化ossd1、卷叶roc5、抽穗期hd1等等,但用于改良不育系包颈遗传障还没有报道。
技术实现要素:
本发明针对现有技术的不足,提供一种基于crispr/cas9系统定点突变水稻中催化赤霉素降解的氧化酶编码基因eui,使得eui基因编码的酶蛋白失活,导致水稻在抽穗时最上节间积累大量赤霉素,引起节间剧烈伸长,从而解除水稻雄性不育系包颈遗传障碍的方法,利用crispr/cas9技术定点编辑eui基因,培育获得不带转基因成分且能保持水稻原优良性状的不包颈不育系方法,具有靶向效率高、育种周期短、成本低、实用性强等优势。
为了实现上述目的,本发明所采用的技术方案如下:
1)靶位点的选择:根据籼稻和粳稻eui基因第二外显子中相同的编码序列(seqidno.1)设计两条基于pcrispr/cas9的靶标序列t1(seqidno.2)和t2(seqidno.3)。
上述靶序列为eui基因编码序列中具有5’-(n)nngg-3’特征的核苷酸序列,其中n代表19-20bp的核酸序列,n为a,t,g,c中的任意一个碱基。
2)靶双链接头引物合成:根据靶标序列及酶切位点信息,设计带粘性末端的接头引物,合成tl正向寡核苷酸链tpl-f(seqidno.4)和与之互补的反向寡核苷酸链tpl-r(seqidno.5),t2正向寡核苷酸链tp2-f(seqidno.6)和与之互补的反向寡核苷酸链tp2-r(seqidn0.7)。
3)含有靶标序列中间表达盒载体的构建:将两对靶引物通过引物退火方法合成双链靶序列,用t4dnaligase将双链靶序列连接到经bsai酶切过的pu3-grna和pu6a-grna载体上,构建pu3-t1-grna,
pu6a-t2-grna中间表达盒载体。
4)中间表达盒扩增:以步骤3)连接产物为模板,用引物u-f/靶接头反向引物(反应1),和靶接头正向引物/grna-r(反应2),进行第一轮pcr扩增,再以第一轮pcr两个反应混合产物为模板,分别用两对位置特异表达盒引物u3-f/u3-r和u6a-f/u6a-r进行第二轮pcr扩增。
5)pcrispr/cas9-eui-grna表达载体构建:将步骤4)得到的两个扩增产物大致等量混合,用pcr产物试剂盒纯化,取适量纯化后产物和pylcrispr/cas9质粒,在反应体系中加入bsai内切酶和t4dna链接酶,采用变温边酶切边连接的方法获得含有双靶点pylcrispr/cas9-eui-grna表达载体(如图)。
6)表达载体农杆菌转化及鉴定:将含有双靶点的表达载体pylcrispr/cas9-eui-grna通过电激转化农杆菌eh105,获得含pylcrispr/cas9-eui-grna质粒的农杆菌。提取农杆菌质粒,取约2ng质粒为模板,用第一个靶点的正向引物与第二个靶点的反向引物配对(tpl-f/tp2-r)进行pcr确认。
7)水稻的遗传转化:以培矮64s的愈伤组织作为遗传转化的受体材料,利用农杆菌介导法将pylcrispr/cas9-eui-grna表达载体转入水稻愈伤组织中,并在含有潮霉素的培养基上进行筛选、分化、生根培养,获得转基因t0代植株。
8)转基因阳性株的筛选:提取t0代植株的基因组dna,用载体pylcrispr/cas9特异引物hyg-f/hyg-r进行pcr扩增,能扩增出697bp目的片段的植株为转基因阳性植株。
9)靶位点的检测分析:在水稻eui基因靶位点两侧设计检测引物p-t-f1和p-t-r1,扩增t0代阳性株的目的条带,将目的条带纯化回收,连接t载体进行测序,比较转基因植株和野生型植株序列,确定是杂合体、双等位突变体、纯合突变体或未发生突变的单株。
10)t1代无选择标记基因突变株的获得:选取t0代纯合突变体或双等位突变体株,低温条件下自交繁殖获得到t1代。以t1代转化植株叶片基因组dna为模板,再用潮霉素抗性基因特异性引物进行pcr扩增用,不能扩增出目的条带的植株为无选择标记基因的纯突变株。
与现有技术相比,本发明的优点在于:
(1)本发明提供了一种基于crispr/cas9系统靶向突变eui解除水稻籼型不育系包颈遗传障碍的方法。eui是编码催化ga分解代谢的基因,该基因发生突变可导致水稻抽穗时最上节间中积累大量的ga,从而引起最上节间的剧烈伸长,克服杂交水稻不育系稻穗不能完全伸出剑叶叶鞘的包穗现象。本发明基于crispr/cas9系统,定点突变eui,快速靶向改良水稻包颈遗传障碍,获得不带转基因成分、穗颈节完全伸出叶鞘的不育系材料。这种技术方法一方面规避免了转基因可能带来的安全隐患,另一方面由于是靶向突变,具有突变位点精准可控的优点。
(2)本发明提供了一种基于crispr/cas9系统定点突变eui获得隐性不包颈不育系的方法,大大缩短了育种周期,加快了育种进程,节省了时间和成本。
(3)本发明提供的基于crispr/cas9系统定点突变eui培育吐颈不育系方法,既避免了杂交、回交法难以完全保留原受体材料遗传背景的弊端,又克服了直接诱变法缺乏定向性和有益突变频率极低的缺点,能够快速、精确定向改良不育系包颈遗传障碍,对于提高杂交水稻制种产量和质量、降低种子生产成本以及防止赤霉素污染环境具有重要意义。
(4)本发明提供了一种基于crispr/cas9系统靶向突变eui培育不包颈不育系的方法,采用5’-(n)n-ngg-3'结构的靶标序列,靶标序列ngg上游的12bp在水稻基因组中有较好的特异性,能显著降低脱靶概率。
附图说明
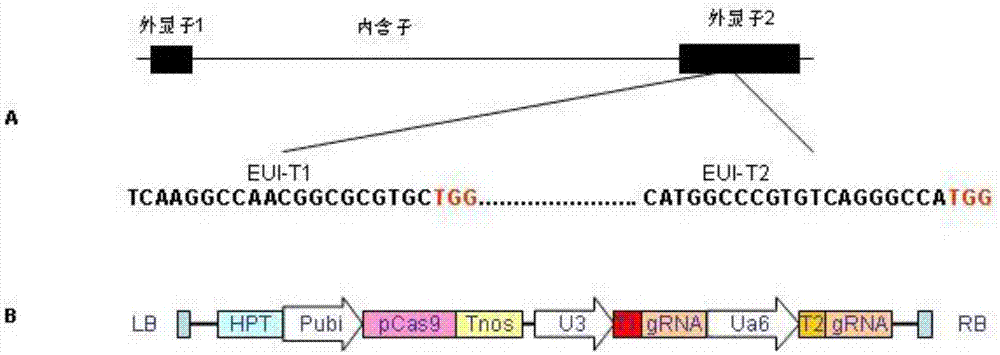
图1为实施例1中pylcrispr/cas9-eui-grna构建示意图,a:eui基因结构和靶序列;b:pylcrispr/cas9-eui-grna构建图
图2为实施例3中的t0代阳性植株的筛选;其中1-8表示8株不同的t0代植株,ck十表示阳性对照,ck一表示阴性对照。
图3为实施例3中的ti代敲除株系hptpcr检测结果;其中1-12表示12株不同的t1代植株,ck十表示阳性对照,ck一表示阴性对照。
图4为实施例3中的突变体eui和对照单株、单茎、节间长度比较。
具体实施方式
下面结合附图以及具体实施方式,对本发明做进一步说明。
实施例1,基于pylcrispr/cas9系统水稻eui基因定点敲除表达载体的构建
(1)基因组靶点选择;从国家水稻数据中心(http://www.ricedata.cn/gene/)查询eui基因座编号(loc_os05g40384),点击基因座编号进入(http://rice.plantbiology.msu.edun/)网查询eui基因结构,如图1a所示,该基因由两个外显子和一个内含子组成,第一外显子长355bp第二外显子长1379bp,内含子长7874bp。再从gramene(http://www.gramene.org/)数据库中下载籼稻(oryzaindica)和粳稻(oryzasativa)eui基因的cds序列,通过序列比较,选择籼稻和粳稻eui基因第二外显子核苷酸序列相同的seqidno.1片段,该片段用于翻译起始密码子atg后第356一1018位的核普酸序列;利用生物信息学网站(http://crispr.dbcls.jp/)软件在seqidno.1序列中查找具有5’-(n)n-ngg-3’(n=19或20,n为a、t、c、g特征序列,从查找到的序列中选靠近pam区特异性较好的tl(seqidno.2)和t2(seqidno.3)两条作为靶标序列。seqidn0.1及两条作为靶标序列序列如下:
其中加粗部分为选择的两条靶序列,阴影部分为pam。
(2)靶双链接头引物合成;根据选定的靶标序列设计与之互补的反向靶序列,并在正、反靶标序列5’添加用于连接到载体的粘性末端接头引物。按照靶标及接头引物序列合成tl正向寡核苷酸链tpl-f(seqidno.4)和与之互补的反向寡核苷酸链tpl-r(seqidno.5),t2正向寡核苷酸链tp2-f(seqidno.6)和与之互补的反向寡核苷酸链tp2-r(seqidn0.7),具体序列为:
tpl-f(seqidno.4):ggcatcaaggccaacggcgcgtgc;
tpl-r(seqidno.5):aaacgcacgcgccgttggccttga;
tp2-f(seqidno.6):ggcgcatggcccgtgtcagggcca;
tp2-r(seqidno.7):aaactggccctgacacgggccatg.
其中,大写字母为靶标序列tl(seqidno.2),t2(seqidn0.3)中去除ngg的序列或其互补序列,小写字母为用于连接载体的粘性末端。
(3)含有靶标序列中间载体表达盒的构建;将两对靶引物通过引物退火方法合成双链靶序列,用t4dnaligase将双链靶序列连接到经bsai酶切过的pu3-grna和pu6a-grna载体上,构建pu3-t1-grna,pu6a-t2-grna中间载体表达盒。
(4)pylcrispr/cas9-eui-grna表达载体构建;4.1表达盒扩增:以步骤3连接产物为模板,用引物u-f/靶接头反向引物(反应1),和靶接头正向引物/grna-r(反应2),进行第一轮高保真pcr扩增,再以第一轮pcr两个反应混合产物为模板,分别用两对位置特异表达盒引物u3-f/u3-r和u6a-f/u6a-r进行第二轮高保真pcr扩增。把得到的两个产物大致等量混合,用pcr产物试剂盒纯化。扩增所用引物的序列分别为:
u-f(seqidno.8):ctccgttttacctgtggaatcg;
grna-r(seqidno.9):cggaggaaaattccatccac
u3-f(seqidno.10):ttcagaggtctctctcgcactggaatcggcagcaaagg;
u3-r(seqidno.11):agcgtgggtctcgtcagggtccatccactccaagctc;
u6a-f(seqidno.12):ttcagaggtctctctgacactggaatcggcagcaaagg;
u6a-r(seqidno.13):agcgtgggtctcgaccgggtccatccactccaagctc;
4.2表达载体构建:将上述得到的两个扩增产物大致等量混合,用pcr产物试剂盒纯化,取步纯化后产物约30ng,加入80ngpylcrispr/cas9质粒,在反应体系中加入bsai内切酶和t4dna链接酶,采用变温边酶切边连接的方法获得含有双靶点pylcrispr/cas9-eui-grna表达载体,如图1b所示。
(5)pylcrispr/cas9-eui-grna表达载体根癌农杆菌转化及鉴定;将4.2重组表达载体pylcrispr/cas9-eui-grna通过电激转化农杆菌eh105,获得含有重组表达载体pylcrispr/cas9-eui-grna的重组农杆菌。提取重组农杆菌质粒,取约2ng质粒为模板,用第一个靶点的正向引物与第二个靶点的反向引物配对(tpl-f/tp2-r)进行pcr确认。
实施例2、水稻eui基因定点敲除pylcrispr/cas9表达载体的遗传转化
将实施例1中的重组农杆菌pylcrispr/cas9-eui-grna侵染水稻两系不育系培矮64s成熟胚诱导的愈伤组织,将获得的水稻转化植株分别命名为l64s,实验具体方法如下:
(1)成熟胚愈伤组织诱导:将水稻种子去壳后先用水洗干净,放在75%乙醇中消毒5min,再放在33%次氯酸钠中灭菌30-45min。用无菌水洗干净种子,去除次氯酸钠,洗到没有明显味道为止。将种子置于nbd2(2,4-d浓度为2mgl-1)培养基平板上,于260c下暗培养10-14d。黄色愈伤组织长出后,用刀片切下诱导出来的新愈伤组织,在新的nbd2培养基上培养l0d后用于转化。
(2)农杆菌的活化:挑取含有pylcrispr/cas9-eui-grna重组农杆菌eha105单菌落,在yeb培养基中震荡培养过夜到od600为0.8左右,8000×g离心5min,用aam-as(as浓度为200μmoll-1)液体培养基重新悬浮沉淀至od600值为0.3-0.6。
(3)农杆菌的转化:将愈伤组织放入农杆菌悬液中20min,轻柔震荡。取出愈伤组织,用灭菌的吸水纸或纱布吸干水分,放在nbd2-as(as浓度为100μmoll-1)培养基的平板上,于28℃下暗培养3d。
(4)抗性愈伤组织的筛选:取出愈伤组织,于滤纸上晾干后放在含有40mgl-1潮霉素的nbd2培养基平板上筛选10d,筛选2次后将新长出的愈伤组织放在分化培养基平板(潮霉素20mgl-1)上,光下培养10d,直到幼苗长出。
(5)转基因植株的再生:将无根幼苗移入含有生根培养基(1/2ms)的试管中,诱导生根。当根形成后,把苗移入温室种植。
实施例3、水稻eui基因定点敲除的检测及不带转基因成分长穗颈不育系的获得
(1)阳性植株鉴定:取实施例2得到的t0代植株叶片,用ctab法提取基因组dna,并以此为模板,用潮霉素抗性基因hpt的特异性引hpt-f/hpt-r进行pcr扩增,pcr产物经琼脂糖凝胶电泳检测潮霉素抗性基因hpt标记,t0代转化的阳性植株基因组dna能扩增出697bp的特异性条带,非阳性植株不能扩增出目的条带如图2所示。
hpt-f(seqidno.14):atttgtgtacgcccgacagt
hpt-r(seqidno.15):gtgcttgacattggggagtt
图3为实施例3中的t0代敲除株系hptpcr检测结果;其中1-8表示12株不同的t1代植株,+表示阳性对照,-表示阴性对照。从图2中可知hpt在tl代群体发生了分离,株系2,6,8不含hpt。
(2)突变位点的检测:以靶标位点为中心,在两侧各离开100-300bp处设计特异性引物p-t-f1和p-t-r1,用高保真酶扩增t0代阳性株和野生型的目的条带,将目的条带纯化回收,连接t载体进行测序,比对转基因植株与野生型植株测序结果,确定是杂合体、双等位突变体、纯合突变体或未发生突变的单株。
p-t-f1(seqidno.16):tgtacgtgacggacccggagct;
p-t-r1(seqidno.17):gcgaagggatgctgaagatga。
表1crispr/cas9系统诱导的t0代转基因植株突变序列
表1为实施例3中的crispr/cas9系统引入的t0代转基因植株突变序列分析;1-5表示不同的转基因株系,ck表示对照;wt表示野生型;阴影字母为pam序列;“一”表示碱基缺失,“+”表示碱基插入,其后的数字表示碱基数。
(3)不带转基因成分突变体筛选:根据测序比对结果,选取t0代纯合突变株系(如表1中的1,4株系)或双等位突变体株,低温条件下自交繁殖获得到t1代。用ctab法提取水稻t1代转化植株叶片基因组dna,并以此为模板,再用潮霉素抗性基因特异性引物进行pcr扩增,不能扩增出目的条带的植株为无选择标记基因的纯突变株。
图2为实施例3中的t1代敲除株系hptpcr检测结果;其中1-12表示12株不同的t1代植株,ck+表示阳性对照,ck-表示阴性对照。从图2中可知hpt在tl代群体发生了分离,株系2,6,8不含hpt。
靶位点的检测分析:在水稻eui基因靶位点两侧设计检测引物p-t-f1和p-t-r1,扩增t0代阳性株的目的条带,将目的条带纯化回收,连接t载体进行测序,比较转基因植株和野生型植株序列,确定是杂合体、双等位突变体、纯合突变体或未发生突变的单株。
(4)突变植株t1代表型观察分析:t1代不带转基因成分的纯合突变株系l64-s在抽穗开花前与对照培矮64s没有明显的差异,但到抽穗开花时极显著高于培矮64s,如下表2所示,穗颈节完全伸出叶鞘,不存在包穗现象,而培矮64s约有1/4-1/3穗被包于剑叶叶鞘中。突变株系株高的增加主要由于倒1节和倒2节显著伸长,而倒3、倒4节间伸长不显著。其他的生物学性状如穗粒大小、每穗总粒数、生物产量等无显著差异。结果表明,通过eui基因编辑得到的突变株系可以使穗包颈障碍得到彻底改善,而其他主要经济性状与原材料没有显著差异。
表2突变株系l64s与培矮64s农艺性状比较
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制。虽然本发明已以较佳实施例揭示如上,然而并非用以限定本发明。任何熟悉本领域的技术人员,在不脱离本发明的精神实质和技术方案的情况下,都可利用上述揭示的方法和技术内容对本发明技术方案做出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同替换、等效变化及修饰,均仍属于本发明技术方案保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!