一种微波促进N-苯磺酰基硝基苯胺类化合物合成的方法与流程
本发明涉及化学合成技术;更具体地,本发明涉及一种微波促进n-苯磺酰基硝基苯胺类化合物合成的方法。
背景技术
研究发现脱落酸能够增强植物体内过氧化物酶、过氧化氢酶活性,积累脯氨酸等进而提高植物的逆境耐受性,因此在生产、生活中具有广泛的应用前景如抗旱、抗寒、耐盐以及耐重金属等等。然而,脱落酸在实际生产、生活中的应用受限于以下几点:(1)小分子侧链末端的碳碳双键因易发生光异构化而失活;(2)在体内p450酶的催化作用和后续反应转化为完全丧失生物活性的物质;(3)合成步骤多、难度大、收率:低、成本高等等。因此,寻找生物活性更高的、性质更稳定的、合成更简单的、综合性能更优越的脱落酸类似物成为了当前研究的热点。2009年,国际顶级期刊science报道了pyr/pyl/rcacs能与脱落酸发生特异性结合并诱导下游相应信号,最终确定了两者的结合机制,以此为基础借助计算机辅助首次发现了一种结构完全不同于脱落酸的具有芳基磺酰胺片段的受体激动剂pyrabactin,但是该小分子活性并不理想,具有进一步改造和优化的潜力。先前我们从pyrabactin出发,通过结构优化找到了一类含硝基的磺酰胺化合物其抗逆活性明显优于脱落酸具有进一步研究和开发的价值,详见cn107372504a和cn107382842a(申请公开号)。此外,n-苯磺酰基硝基苯胺类化合物还是医、农药的重要中间体,具有广泛的生物活性,并且一般采用先将取代苯胺与苯磺酰氯作用得到n-苯磺酰基苯胺,再经浓硫酸和浓硝酸混酸进行硝化反应的方法进行合成,该方法产生大量废酸易造成环境污染。
因此,开发一种快速、高效、低污染的n-苯磺酰基硝基苯胺类化合物合成的方法具有广泛的应用价值和经济价值,此外将其用于提高植物抗逆性具有极大潜力。
技术实现要素:
通过对n-苯磺酰基硝基苯胺类化合物合成方法的研究,本发明提供了一种微波辐射的、苯磺酰胺和卤代硝基苯为原料的快速合成n-苯磺酰基硝基苯胺类化合物的方法,减少了步骤,避免了硝化带来的环境问题,极大地降低了污染,同时该n-苯磺酰基硝基苯胺类化合物能够用于提高植物抗逆性。
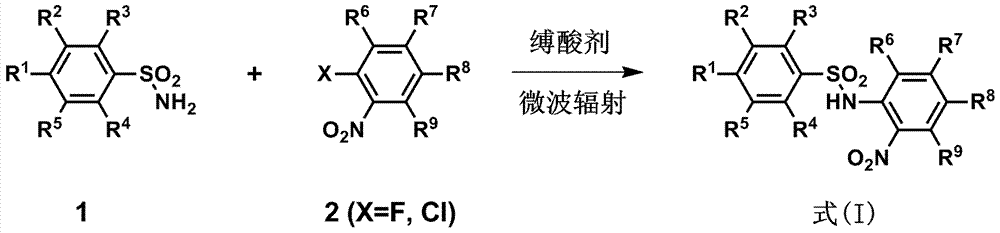
为了实现上述目的,本发明提供了一种微波辅助的、苯磺酰胺和卤代硝基苯为原料,高效、高收率合成方法,该n-苯磺酰基硝基苯胺类化合物具有式(i)所示的结构,
其中,在式(ⅰ)中r1、r2、r3、r4、r5、r6、r7、r8和r9分别选自h、c1-c4的烷基、c1-c4的烷氧基、硝基、氰基、酯基、三氟甲基、三氟甲氧基和卤素,或者r2与r3形成取代或未取代的芳香环。
一种快速、高效、低污染的n-苯磺酰基硝基苯胺类化合物的合成方法,其主要特征为采用微波辐射的方式,以廉价易得的苯磺酰胺和卤代硝基苯为原料在缚酸剂的作用下制得n-苯磺酰基硝基苯胺类化合物,该合成方法主要特征如下:
其中,在式(ⅰ)中r1、r2、r3、r4、r5、r6、r7、r8和r9分别选自h、c1-c4的烷基、c1-c4的烷氧基、硝基、氰基、酯基、三氟甲基、三氟甲氧基和卤素,或者r2与r3形成取代或未取代的芳香环。
进一步地,在上述技术方案中,微波辐射温度为60-140摄氏度。优选80-140摄氏度。
进一步地,在上述技术方案中,微波辐射时间为5-60分钟。优选10-30分钟。
进一步地,在上述技术方案中,所述缚酸剂选自碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾、碳酸铯、磷酸钾、碳酸锂、氢氧化钾、氢氧化钠等无机缚酸剂,或三乙胺、n-甲基吗啉、n,n-二异丙基乙胺等有机缚酸剂中的一种或多种。优选碳酸钾。
进一步地,在上述技术方案中,所述有机溶剂为1,4-二氧六环,吡咯烷酮,n,n-二甲基甲酰胺,n,n-二甲基乙酰胺,二甲亚砜的一种或多种。优选n,n-二甲基甲酰胺。
进一步地,在上述技术方案中,所述原料苯磺酰胺和卤代硝基苯的摩尔比为1:1-1:1.5,优选摩尔比为1:1.1;原料苯磺酰胺与缚酸剂的摩尔比为1:1-1:3,优选摩尔比为1:1.5。
与现有技术相比,本方面技术方案实施带来的有益效果是:
1、缩短了合成步骤,提高了合成的效率和收率。
2、反应原料来源广泛,取代苯磺酰胺和取代卤代硝基苯均为常见化学试剂。
3、该策略可以用于快速合成式(ⅰ)所示结构的n-苯磺酰基硝基苯胺类化合物。
4、降低了硝化反应带来的环境污染问题。
本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图为说明书的一部分,与下面的具体实施方式一起用于进一步解释本发明,但并不构成对本发明的限制。在附图中:
图1是微波促进n-苯磺酰基硝基苯胺类化合物合成的路线图。
图2是化合物15的1h-nmr谱图。
具体实施方式
下面对本发明的具体实施方式进行详细地说明。应当理解为,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
本发明提供了一种微波辅助的、苯磺酰胺和卤代硝基苯为原料的快速合成n-苯磺酰基硝基苯胺类化合物的方法,该n-苯磺酰基硝基苯胺类化合物具有式(i)所示的结构,
其中,在式(ⅰ)中r1、r2、r3、r4、r5、r6、r7、r8和r9分别选自h、c1-c4的烷基、c1-c4的烷氧基、硝基、氰基、酯基、三氟甲基、三氟甲氧基和卤素,或者r2与r3形成取代或未取代的芳香环。该合成方法如下:
具体实施例1
n-(4-溴苯磺酰基)-2-硝基苯胺(化合物3)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:95%;熔点:124.3-125.1℃;1hnmr(600mhz,dmso)δ11.70(s,1h),7.94(s,4h),7.76(d,j=7.8hz,2h),7.52(t,j=7.8hz,2h)。
具体实施例2
n-(4-溴苯磺酰基)-2-硝基-4-甲基苯胺(化合物4)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-甲基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应20分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:92%;熔点:124.3-125.1℃;1hnmr(600mhz,dmso)δ11.70(s,1h),7.94(s,4h),7.76(d,j=7.8hz,2h),7.52(t,j=7.8hz,2h)。
具体实施例3
n-(4-溴苯磺酰基)-2-硝基-4-甲氧基苯胺(化合物5)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-甲氧基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应25分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:88%;熔点:120.3–121.3℃;1hnmr(600mhz,dmso)δ10.21(s,1h),7.79(d,j=8.4hz,2h),7.54(d,j=8.4hz,2h),7.44(d,j=3.0hz,1h),7.21(dd,j=9.0hz,j=3.0hz,1h),7.05(d,j=9.0hz,1h),3.81(s,3h)。
具体实施例4
n-(4-溴苯磺酰基)-2-硝基-4-氟苯胺(化合物6)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2,5-二氟硝基苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:91%;熔点:127.2-127.9℃;1hnmr(600mhz,dmso)δ10.47(s,1h),7.93(d,j=7.8hz,1h),7.80(d,j=7.8hz,2h),7.57(m,3h),7.23(m,1h)。
具体实施例5
n-(4-溴苯磺酰基)-2-硝基-4-氯苯胺(化合物7)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-氯氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:92%;熔点:120.3-121.2℃;1hnmr(600mhz,cdcl3)δ9.74(s,1h),8.12(d,j=2.4hz,1h),7.82(d,j=9.0hz,1h),7.70(d,j=9.0hz,2h),7.63(d,j=9.0hz,2h),7.57(dd,j=9.0,2.4hz,1h)。
具体实施例6
n-(4-溴苯磺酰基)-2-硝基-4-溴苯胺(化合物8)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-溴氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应20分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:93%;熔点:127.3-128.1℃;1hnmr(600mhz,cdcl3)δ9.79(s,1h),8.28(s,1h),7.76(d,j=9.0hz,1h),7.71(d,j=9.0hz,3h),7.64(d,j=8.4hz,2h)。
具体实施例7
n-(4-溴苯磺酰基)-2-硝基-4-三氟甲基苯胺(化合物9)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-三氟甲基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应10分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:92%;熔点:137.1-137.9℃;1hnmr(600mhz,dmso)δ10.95(s,1h),8.30(s,1h),8.02(d,j=8.4hz,1h),7.84(d,j=8.4hz,2h),7.76(d,j=8.4hz,2h),7.56(d,j=8.6hz,1h)。
具体实施例8
n-(4-溴苯磺酰基)-2-硝基-4-三氟甲氧基苯胺(化合物10)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-三氟甲氧基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:91%;熔点:99.2-100.1℃;1hnmr(600mhz,dmso)δ10.68(s,1h),8.04(s,1h),7.83(d,j=8.4hz,2h),7.72(d,j=8.4hz,1h),7.67(d,j=7.8hz,2h),7.39(d,j=9.0hz,1h)。
具体实施例9
n-(4-溴苯磺酰基)-2-硝基-4-氰基苯胺(化合物11)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-氰基氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应10分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:94%;熔点:192.6-193.3℃;1hnmr(600mhz,dmso)δ8.47(s,1h),8.02(d,j=8.4hz,1h),7.83(d,j=8.4hz,2h),7.76(d,j=9.0hz,2h),7.50(d,j=8.4hz,1h)。
具体实施例10
n-(4-溴苯磺酰基)-2-硝基-4-溴-6-氯苯胺(化合物12)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2-硝基-4-溴-6-氯氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:89%;熔点:177.3–178.2℃;1hnmr(600mhz,dmso)δ10.91(s,1h),8.23(s,1h),8.17(s,1h),7.78(d,j=8.4hz,2h),7.54(d,j=8.4hz,2h)。
具体实施例11
n-(4-溴苯磺酰基)-2-硝基-4-氯-6-氟苯胺(化合物13)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2,3-二氟-5-氯硝基苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:87%;熔点:118.1-118.8℃;1hnmr(600mhz,cdcl3)δ8.26(s,1h),7.85(s,1h),7.68(q,j=9.0hz,4h),7.47(dd,j=9.6,2.4hz,1h)。
具体实施例12
n-(4-溴-1-萘磺酰基)-2-硝基-4-氟苯胺(化合物14)的合成:将1mmol4-溴-1-萘磺酰胺与1.1mmol2,5-二氟硝基苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:89%;熔点:142.6-143.4℃;1hnmr(600mhz,dmso)δ10.80(s,1h),8.62(d,j=8.4hz,1h),8.34(d,j=8.4hz,1h),8.02(d,j=7.8hz,1h),7.89(d,j=7.8hz,1h),7.88–7.84(m,2h),7.81(t,j=7.8hz,1h),7.47(td,j=9.0,3.0hz,1h),7.11(dd,j=9.0,5.4hz,1h)。
具体实施例13
n-(4-溴-1-萘磺酰基)-2-硝基-4-氯苯胺(化合物15)的合成:将1mmol4-溴-1-萘磺酰胺与1.1mmol2-硝基-4-氯氟苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:90%;熔点:160.1-161.1℃;1hnmr(600mhz,cdcl3)δ10.24(s,1h),8.63(d,j=8.4hz,1h),8.38(d,j=8.4hz,1h),8.17(d,j=7.8hz,1h),8.02(d,j=2.4hz,1h),7.87(d,j=7.8hz,1h),7.80–7.70(m,3h),7.47(dd,j=9.0,2.4hz,1h)。
具体实施例14
n-(4-溴苯磺酰基)-2,6-二硝基-4-甲基苯胺(化合物16)的合成:将1mmol4-溴苯磺酰胺与1.1mmol2,6-二硝基-4-甲基氯苯加入含有1.5mmol无水碳酸钾和5mln,n-二甲基甲酰胺(dmf)反应管中,随后盖好反应管盖子,140摄氏度下微波辐射反应15分钟后冷却至室温,打开后酸化,乙酸乙酯萃取后合并有机相,无水硫酸钠干燥后,滤液减压脱干,柱层析,洗脱剂为石油醚和丙酮。得黄色固体,收率:91%;熔点:203.3-203.9℃;1hnmr(600mhz,dmso)δ11.04(s,1h),8.11(s,2h),7.76(d,j=8.4hz,2h),7.46(d,j=8.4hz,2h),2.45(s,3h).
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对技术方案进行多种简单的变型,这些简单的变型均属于本发明的保护范围。
另外需要说明的是,如上具体实施方式所描述的各具体技术特征,在不矛盾的情况下,可通过合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
- 还没有人留言评论。精彩留言会获得点赞!