一种单细胞DNA文库的构建方法与流程
本发明涉及生物
技术领域:
中,一种单细胞dna文库的构建方法。
背景技术:
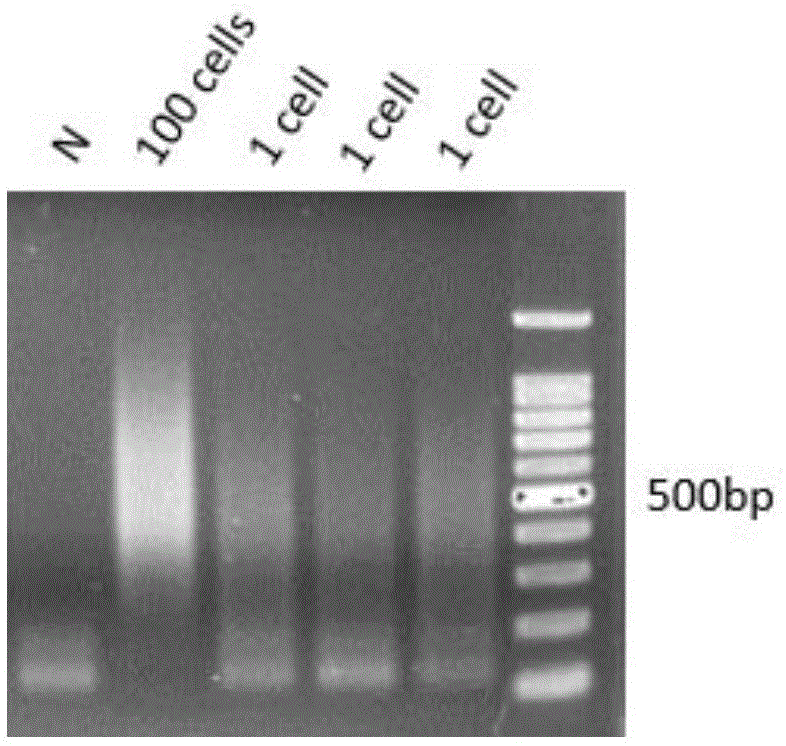
:多细胞基因组的测序中,对来源于混合细胞的dna进行高深度的测序。这类方法能够鉴定主要的细胞亚克隆,但受测序错误的限制,对少数细胞群体的鉴定能力弱。多细胞的测序方法会受到细胞数目的限制,且对亚克隆细胞中低频率的dna拷贝数变异(cnv)和单核苷酸变异(snv)的检测不灵敏。单细胞基因组学将基因组的研究延伸到了细胞水平,为对遗传学的理解提供新的观点。单细胞基因组学可用于鉴定和组装不可培养的微生物的基因组,评估遗传镶嵌在正常生理状态和疾病状态下的作用,以及确认肿瘤内的遗传异质性在癌症发展和治疗反应中所起到的作用,单细胞基因组学对珍贵的细胞样品,如胚细胞、循环肿瘤细胞的研究具有重要意义。现有的单细胞基因组技术,主要依赖于测序文库构建前的单细胞全基因组扩增来获得完整的基因组信息,主要有简并寡核苷酸引物pcr、多重置换扩增、dlp直接文库构建、lianti技术和tnbc技术。简并寡核苷酸引物pcr(degenerateoligonucleotide-primedpcr(dop-pcr)),利用随机引物对单细胞全基因组进行pcr预扩增,增大起始的dna量,再进行后续的文库构建。dop-pcr需要预扩增,会偏好性地扩增基因组上胞嘧啶和鸟苷含量丰富的区域,导致较低的基因组物理覆盖度,不利于单核苷酸变异snv的检测。多重置换扩增(multipledisplacementamplification(mda)),利用随机引物在具有合成能力和链置换活性的聚合酶作用下,对单细胞的基因组dna进行预扩增,增大起始的dna量,然后进行文库构建。该方法覆盖度较好,具有较低的错误率,假阳性较低,但均一性较差。该方法需要预扩增,扩增均一性较差,且φ29聚合酶的活性会产生少量嵌合序列副产物,不利于拷贝数变异cnv的检测。dlp直接文库构建(directlibrarypreparation(dlp)),利用tn5酶转座活性直接处理原始单细胞dna,在定制的微流控仪器上完成文库构建。其中,由于nextera的tn5酶的引物设计使用两种不同的接头,只有50%的基因组能够被扩增、测序。此外,该技术需要定制微流控的设备,预先在相应的孔中固定特定的引物,设计较复杂,不利于该技术的应用。lianti技术(linearamplificationviatransposoninsertion(lianti)技术),利用对称性接头打断单细胞基因组dna,提高转座酶模板利用率,并使用t7promoter进行转录再逆转录成cdna,实现线性扩增降低偏好性和pcr错误,可大幅提升单细胞dna测序的cnv分辨率和碱基准确性。该方法未检测插入片段的大小是否适合于后续的二代测序,片段长度过长会影响测序的覆盖度。此外,转录和反转录的过程时间较长,成本较高。tnbc技术(transposonbarcoded(tnbc)技术),利用饱和浓度的mu转座酶打断原始单细胞dna,再用与mu匹配的muend引物进行扩增纯化,纯化后的产物以带有barcode的引物进行文库构建。进行二代测序后,比对到参考基因组,将每个片段起始和末位的几个碱基作为独特片段标记(ufi)。根据ufi去除冗余的序列、连接各dna片段,以及构成单倍型。该方法经过mu转座酶打断扩增后,需纯化后再进行带有barcode的引物扩增,易在操作过程中损失部分基因组信息。技术实现要素:本发明所要解决的技术问题是如何构建单细胞全基因组文库。为解决上述技术问题,本发明首先提供了一种单细胞全基因组文库的构建方法,所述方法包括:利用转座酶复合体打断单细胞基因组dna,使打断的片段两端的接头相同,得到片段化基因组,利用所述片段化基因组构建单细胞全基因组文库;所述转座酶复合体包括转座酶和转座酶接头,所述转座酶接头由名称分别为a和c的单链dna组成;所述a为所述转座酶的识别序列的反向互补序列;所述c为含有所述识别序列的单链dna。上述方法中,所述c可由所述识别序列与名称为c1的单链dna组成;所述利用所述片段化基因组构建单细胞全基因组文库包括:利用名称为引物1的引物对所述片段化基因组进行扩增,得到pcr产物,所述pcr产物即为单细胞全基因组文库;所述引物含有所述c1,能识别所述c。所述c1和所述识别序列可分别位于所述c的5′端和3′端。所述转座酶复合体具体可由所述转座酶和所述转座酶接头形成的复合体。所述转座酶复合体具体可由所述转座酶和所述转座酶接头在25℃孵育60min得到。所述识别序列具体为所述转座酶实现转座所依赖的特定的19bpme(mosaicend)序列。在利用所述转座酶复合体打断所述样本基因组dna后,所述片段化基因组的两端分别连接有所述转座酶接头。上述方法中,所述引物1可由所述c中所述c1及所述识别序列的部分或全部序列组成。上述方法中,所述c1可为序列表中序列1的第1-15位。上述方法中,所述转座酶可为tn5。所述识别序列可为序列表中序列1的第16-34位。具体的,所述c的序列可为序列表中序列1。所述引物1的序列具体可为序列表中序列1的第1-22位。上述方法还可包括对所述pcr产物连接用于测序的接头步骤。上述方法还可包括:对所述pcr产物连接所述用于测序的接头,得到连接接头的文库;利用识别所述用于测序的接头的引物对所述连接接头的文库进行pcr扩增,得到单细胞全基因组文库。上述方法中,所述用于测序的接头可为用于高通量测序的接头。所述高通量测序具体可为利用illuminahiseq的高通量测序。所述用于测序的接头具体可为由序列表中序列2和序列3所示的单链dna组成的y型接头。所述识别所述用于测序的接头的引物可含有标签序列(barcode序列)。所述标签序列的长度可为b1)或b2):b1)6-16nt;b2)10nt。所述标签序列在特定的样本中可为确定的序列,以区分特定样本与其他样本的基因组;所述标签序列在特定的样本中也可为不确定的序列,即其序列为4n种,n为标签序列长度,以在测序过程中对该样本的基因组dna进行区分。所述识别所述用于测序的接头的引物具体可为由p5和p7组成的引物对。本发明还提供了下述x1)-x4)中任一产品:x1)所述转座酶复合体;x2)所述转座酶接头;x3)成套试剂,由x31)和x32)组成;x31)所述转座酶复合体或所述转座酶接头;x32)所述引物1;x4)成套试剂,由x41)、x42)和x43)组成;x41)所述转座酶复合体或所述转座酶接头;x42)所述引物1;x43)所述识别所述用于测序的接头的引物。x3)和x4)所述成套试剂均可用于构建单细胞全基因组文库或用于单细胞全基因组测序。本发明还提供了下述任一应用:ⅰ、所述单细胞全基因组文库的构建方法在基因组测序中的应用;ⅱ、所述产品在基因组测序或构建基因组文库中的应用。上述应用中,所述基因组测序可为对单细胞的基因组进行的测序。所述基因组文库可为用于单细胞的基因组文库。本发明以皮克级别的单细胞基因组dna(<10pg)为模板,在无预扩增的条件下利用只包埋一种接头的转座酶打断基因组,使打断的片段两端的接头相同,提高基因组的覆盖度,实现单细胞全基因组文库构建,减少了pcr扩增的偏向性,提高基因组覆盖均一性和覆盖度;本发明单细胞裂解、扩增等过程反应体系简单可以相互兼容,提高了实验的成功率,降低了测序的成本;实验过程简便、耗时短,减少了样品的损失,使测序信息更完整,适合于扩大规模实现高通量的单细胞扩增,优于lianti和tnbc的方法。此外,在测序前扩增又在序列中引入了特异性的barcode,可将多个样品pooling到一起再进行低深度的测序,对dna的拷贝数变异检测灵敏度高、耗时短(整个文库构建过程可在5-6h内完成),精简了单细胞dna预扩增的过程,在单管单细胞层面实现快速的单细胞全基因组文库构建,适用于发现细胞的亚克隆。本发明还可有效结合流式细胞分选技术和高通量自动化操作平台,获得大量单细胞基因组拷贝数变异信息,应用范围广泛。附图说明图1为pcr产物的电泳检测结果。n为阴性对照,100cells表示阳性对照,三个1cell分别为实验组的三个重复。图2为通过agilent2100检测片段分布情况。纵坐标为荧光强度。图3为两种构建文库方法的覆盖度的比较。单接头表示本发明的单细胞dna文库的构建方法构建的文库,双接头表示利用双接头方法构建的文库。图4为两种构建文库方法的测序深度的比较。单接头表示本发明的单细胞dna文库的构建方法构建的文库,双接头表示利用双接头方法构建的文库。具体实施方式下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂、仪器等,如无特殊说明,均可从商业途径得到。以下实施例中的定量试验,均设置三次重复实验,结果取平均值。下述实施例中,如无特殊说明,序列表中各核苷酸序列的第1位均为相应dna的5′末端核苷酸,末位均为相应dna的3′末端核苷酸。实施例1、单细胞dna文库的构建本实施例提供了一种单细胞dna文库的构建方法,该方法利用tn5转座酶包埋含有扩增序列和tn5识别序列的接头,将单细胞的基因组dna(<10pg)在无预扩增的情况下,随机打断并引入同一种接头。用引入的接头引物进行无偏向性地扩增,富集dna。纯化后通过加a和连接反应,在dna片段两端引入illumina文库构建的包含p5、p7序列的y型接头,磁珠纯化后用相应的p5和p7引物扩增,p7引物中带有特异性的barcode,纯化后即为illuminahiseq平台的测序文库。具体操作方法如下:一、转座酶复合体的制备1、合成名称分别为a和c的单链dna,c由名称为c1的扩增接头和转座酶tn5的识别序列(识别位点)组成,二者直接相连,a的序列为转座酶tn5的识别序列的反向互补序列。人工合成寡核苷酸时,其5′端核苷酸不含有磷酸,此处需要在其5′端进行磷酸修饰,以使5′末端核苷酸可以与3′末端核苷酸连接。各引物如下:a:5′-pho-ctgtctcttatacacatct-3′;c:5′-gtctcgtgggctcggagatgtgtataagagacag-3′(序列表中序列1),下划线所示核苷酸序列为c1,加粗核苷酸序列为转座酶tn5的识别序列。2、使用annealingbuffer溶解a和c,浓度均为100μm,其中,annealingbuffer由1mtris-hcl溶液ph7.8(t2694-1l,sigma)、0.5medta溶液ph8.0(am9260g,ambion和5mnacl氯化钠水溶液(s5150-1l,sigma)配制,1lannealingbuffer中含有10ml1mtris-hcl溶液ph7.8,2ml0.5medta溶液ph8.0和10ml5mnacl氯化钠水溶液,余量为水。配制如下反应体系:将上述反应体系涡旋震荡充分混匀,并短暂离心使溶液回到管底。置于pcr仪内,按照如下反应程序进行反应:循环数温度时间175℃15min160℃10min150℃10min140℃10min125℃30min反应结束后混匀,即得到tn5接头,-20℃保存。3、tn5接头包埋a)在灭菌pcr管中依次添加各反应组分:成分体积步骤2得到的tn5接头12.5μltn5转座酶(bgi产品bge005)87.5μl总体积100μlb)使用移液器轻轻吹打,充分混匀。c)将反应置于25℃反应60min,即得到转座酶复合体(命名为tagmentenzymeadvancedmixv5s),置于-20℃存。二、单细胞dna文库的构建1、单细胞裂解液制备取87μl30mmtris-hcl(ph7.5)和3μl蛋白酶(qiagen,catno./id:19155qiagenprotease(7.5au))混合即得到的单细胞裂解液,蛋白酶在该溶液中的浓度为0.67mg/ml。2、单细胞样品制备及裂解将hela细胞用pbs洗涤去除外部dna和细胞碎片,使用流式细胞仪分选单个细胞置入灭菌的装有2μl步骤1的单细胞裂解液的pcr管中,即实验组。以无单细胞的单细胞裂解液作为阴性对照,向单细胞裂解液中添加100个hela细胞作为阳性对照。将实验组、阴性对照和阳性对照于50℃孵育1h进行反应,然后于75℃孵育20min,80℃孵育5min失活蛋白酶,冷却到10℃,即得到单细胞裂解产物(实验组基因组dna含量<10pg)。3、利用tn5酶对单细胞基因组进行片段化a)于室温解冻5xtagbuffer(华大基因产品,货号为bge005b01),上下颠倒混匀后备用;b)按照如下反应体系对单细胞基因组进行片段化:成分体积h205.8μl单细胞裂解产物2μl5xtagbuffer2μl步骤1的转座酶复合体0.2μl总体积10μlc)将上述反应体系于55℃温浴30min,然后向反应体系中添加2.5μl0.1%sds水溶液后室温孵育5min,终止反应,置于冰上,即得到单细胞片段化基因组。按照上述a)-c)的方法,将单细胞裂解产物分别替换为阳性对照或阴性对照进行实验。4、单细胞片段化基因组的pcr扩增按照下述方法,对步骤3得到的单细胞片段化基因组进行pcr扩增:a)引物名称及序列:所用引物为partialcprimer接头引物序列,人工合成寡核苷酸时,其5′端核苷酸不含有磷酸,此处需要在其5′端进行磷酸修饰,以使5′末端核苷酸可以与3′末端核苷酸连接,引物具体如下:partialcprimer:5’-pho-gtctcgtgggctcggagatgtg-3’将灭菌pcr管置于冰浴中,按照如下体系配制pcr扩增体系,其中,5xkapafidelitybuffer和kapahifidnapolymerase均为kapabiotech产品:成分体积灭菌蒸馏水24μl步骤3的单细胞片段化基因组12.5μl5xkapafidelitybuffer10μl10mmeachdntp1.5μlpartialcprimer(10μm)1μlkapahifidnapolymerase(1u/μl)1μl总体积50μl将上述pcr扩增体系按照如下扩增程序进行扩增,得到pcr产物:5、pcr产物的磁珠纯化利用1×ampurexpbead(beckmancoulter)纯化步骤4得到的pcr产物。a)起始pcr产物体积应为50μl。因pcr过程中样品挥发会导致产物体积不足50μl,进行下面操作之前必须使用灭菌蒸馏水将体积补齐至50μl。b)涡旋震荡混匀ampurexpbeads并吸取50μl体积至含有50μlpcr产物的ep管中,使用移液器轻轻吹打10次充分混匀。室温孵育10分钟。c)步骤b)结束后进行短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心移除上清。d)保持ep管始终处于磁力架中,步骤c)结束后,向ep管中加入200μl新鲜配制的80%(体积百分比)乙醇水溶液漂洗磁珠。室温孵育30秒后小心移除上清。e)重复步骤d),总计漂洗两次。f)保持ep管始终处于磁力架中,开盖,空气中干燥磁珠3分钟。g)将ep管从磁力架中取出,向ep管中加入21μl灭菌超纯水,涡旋振荡或使用移液器轻轻吹打充分混匀,将ep管短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心吸取上清至灭菌ep管中,即得到纯化后的pcr产物,于-20℃保存。6、pcr产物末端加a将步骤5获得的纯化后的pcr产物在datp、klenow(exo-3’→5’)酶(enzymatics,货号p7010-hc-l,50,000u/ml)的作用下,添加一个a碱基,用于后续与y型接头的连接。配制如下反应体系:37℃反应30min,4℃保持。其中,10xnebuffer2.1为neb产品,货号为b7202s。7、加a后纯化将步骤6的产物进行柱纯化,纯化过程参考试剂盒purelinkpcrmicrokit(invitrogen,货号a11358),最后用11μleb洗脱产物。8、y型接头制备a)合成名称分别为yp5ad和yp7ad的单链dna,两序列中有部分序列可互补配对,退火后形成y型接头。人工合成寡核苷酸时,其5′端核苷酸不含有磷酸,此处需要在其5′端进行磷酸修饰,以使5′末端核苷酸可以与3′末端核苷酸连接。各引物如下:yp5ad:5’-pho-tacactctttccctacacgacgctcttccgatct-3′(序列2)yp7ad:5’-pho-gatcggaagagcacacgtctgaactccagtcac-3′(序列3)b)使用超纯水溶解yp5ad和yp7ad,浓度均为100μm,在10xnebuffer2.1缓冲液(neb,货号b7202s)的作用下制备y型接头。配制如下反应体系:将上述反应体系涡旋震荡充分混匀,并短暂离心使溶液回到管底。置于pcr仪内,按照如下反应程序进行反应:循环数温度时间175℃15min160℃10min150℃10min140℃10min125℃30min反应结束后混匀,即得到y型接头,-20℃保存。9、y型接头连接利用t4dna连接酶,将步骤7的纯化产物与步骤8获得的y型接头进行连接反应,得到连接产物。反应体系如下:按上述体系配制后,混匀,23℃,60min连接,4℃保持。pnkbuffer:10xpolynucleotidekinasebuffer,enzymatics,货号b904;t4dnaligase(600u/μl),fermentas,货号el0011。10、加接头后磁珠纯化利用1×ampurexpbead(beckmancoulter)纯化步骤9得到的连接产物。a)起始pcr产物体积应为50μl。因pcr过程中样品挥发会导致产物体积不足50μl,进行下面操作之前必须使用灭菌蒸馏水将体积补齐至50μl。b)涡旋震荡混匀ampurexpbeads并吸取50μl体积至含有50μlpcr产物的ep管中,使用移液器轻轻吹打10次充分混匀。室温孵育10分钟。c)步骤b)结束后进行短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心移除上清。d)保持ep管始终处于磁力架中,步骤c)结束后,向ep管中加入200μl新鲜配制的80%(体积百分比)乙醇水溶液漂洗磁珠。室温孵育30秒后小心移除上清。e)重复步骤d),总计漂洗两次。f)保持ep管始终处于磁力架中,开盖,空气中干燥磁珠3分钟。g)将ep管从磁力架中取出,向ep管中加入21μl灭菌超纯水,涡旋振荡或使用移液器轻轻吹打充分混匀,将ep管短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心吸取上清至灭菌ep管中,即得到纯化后的pcr产物,于-20℃保存。纯化后产物取1μl利用qubitdsdnahsassaykit(thermofisherscientific)测定dsdna浓度。11、加接头后扩增用p5(5’-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct)和p7(5’-caagcagaagacggcatacgagataaccgcgggtgactggagttcagacgtgt,加粗部分为barcode可根据不同样品更换带有不同barcode的p7引物)组成的引物对对步骤10获得的纯化后产物进行扩增,以获得充足的illumina测序文库。按如下体系配制:其中,5xkapafidelitybuffer和kapahifidnapolymerase均为kapabiotech产品:成分体积灭菌蒸馏水13.5μl步骤10的纯化产物20μl5xkapafidelitybuffer10μl10mmeachdntp1.5μlp5(amp)(10μm)2μlp7(tag)(10μm)2μlkapahifidnapolymerase(1u/μl)1μl总体积50μl将上述pcr扩增体系按照如下扩增程序进行扩增,得到pcr产物:12、磁珠纯化利用1×ampurexpbead(beckmancoulter)纯化步骤11得到的pcr产物。a)起始pcr产物体积应为50μl。因pcr过程中样品挥发会导致产物体积不足50μl,进行下面操作之前必须使用灭菌蒸馏水将体积补齐至50μl。b)涡旋震荡混匀ampurexpbeads并吸取50μl体积至含有50μlpcr产物的ep管中,使用移液器轻轻吹打10次充分混匀。室温孵育10分钟。c)步骤b)结束后进行短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心移除上清。d)保持ep管始终处于磁力架中,步骤c)结束后,向ep管中加入200μl新鲜配制的80%(体积百分比)乙醇水溶液漂洗磁珠。室温孵育30秒后小心移除上清。e)重复步骤d),总计漂洗两次。f)保持ep管始终处于磁力架中,开盖,空气中干燥磁珠3分钟。g)将ep管从磁力架中取出,向ep管中加入21μl灭菌超纯水,涡旋振荡或使用移液器轻轻吹打充分混匀,将ep管短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心吸取上清至灭菌ep管中,即得到纯化后的pcr产物,于-20℃保存。纯化后产物取1μl利用qubitdsdnahsassaykit(thermofisherscientific)测定dsdna浓度。13.质量与评估a)质量浓度检测利用qubitdsdnahsassaykit(thermofisherscientific)测定,实验组的浓度大于3.5ng/μl。b)电泳检测采用1%琼脂糖凝胶检测纯化后的pcr产物。结果(图1)显示,阴性对照(n)的泳道只有100bp左右的引物二聚体,实验组及阳性对照都有明显的200bp以上的产物条带。c)agilent2100检测将纯化后的pcr产物稀释到合适的浓度,通过bioanalyzerhighsensitivitydnakit(agilent)测定pcr产物片段大小范围。结果(图2)显示,实验组的pcr产物主要集中在300bp,主要在300bp~500bp之间,符合预期的片段大小。14、使用illuminahiseq平台方法进行测序对步骤12得到的纯化产物利用illuminahiseq平台进行测序,测序结果的覆盖度和测序深度(测序深度为测序得到的总核苷酸数与待测基因组大小的比值)分别如图3和图4所示,单接头方法的覆盖度为3.24%,测序深度为0.05;双接头方法的覆盖度为2.00%,测序深度为0.02。上述结果表明,利用单接头构建文库的方法优于利用双接头构建,覆盖度和测序深度都得到大幅度提高。另外,结果显示,实验组的覆盖度和测序深度均与阳性对照组无显著差异。其中,利用双接头构建文库的操作步骤如下:1.接头(adaptermix)制备a)引物名称及序列:primera:5'-[phos]ctgtctcttatacacatct-3'primerb:5'-tcgtcggcagcgtcagatgtgtataagagacag-3'primerc:5'-gtctcgtgggctcggagatgtgtataagagacag-3'b)使用annealingbuffer溶解primera、primerb、primerc至100μm。c)分别配制如下反应体系:注:可根据实际需要等比放大反应体系。d)分别将反应1和反应2涡旋震荡充分混匀,并短暂离心使溶液回到管底。置于pcr仪内,进行如下反应程序(hotlid:105℃):hold温度时间175℃15min160℃10min150℃10min140℃10min125℃30mine)反应结束后,将反应1和反应2的产物等体积混合,混匀。命名为adaptermix,-20℃保存。2.接头(adaptermix)包埋a)在灭菌pcr管中依次添加各反应组分:成分体积adaptermix12.5μltn5转座酶(bgi产品bge005)87.5μl总体积100μlb)使用移液器轻轻吹打,充分混匀。c)将反应置于25℃反应60min。反应产物命名为tagmentenzymeadvancedmixv5s,置于-20℃存。3.单细胞裂解液制备取87μl30mmtris-hcl(ph7.5)和3μl蛋白酶(qiagen,catno./id:19155qiagenprotease(7.5au))混合即得到的单细胞裂解液,蛋白酶在该溶液中的浓度为0.67mg/ml。4.单细胞样品制备及裂解hela细胞用pbs洗涤去除外部dna和细胞碎片,使用流式细胞仪分选单个细胞进入灭菌的装有2μl步骤1的单细胞裂解液的pcr管中。以无单细胞的单细胞裂解液作为阴性对照,向单细胞裂解液中添加100个hela细胞作为阳性对照。50℃1h孵育反应。75℃20min,80℃5min失活蛋白酶,冷却到10℃。5.利用tn5酶对单细胞基因组进行片段化。a)于室温解冻5xtagbuffer(华大基因产品,货号为bge005b01),上下颠倒混匀后备用;确认0.1%sds处于室温,并轻弹管壁确认有无沉淀。如有沉淀,可于37℃加热并剧烈震荡充分混匀沉淀即会溶解。b)在上述包含裂解产物的pcr管中依次添加各反应组分:成分体积h205.8μl裂解产物2μl5xtagbuffer2μlfragmentenzymeadvancedmixv5s0.2μl总体积10μlc)55℃温浴30min,添加2.5μl0.1%sds水溶液室温5min,终止反应,置于冰上。6.片段化产物pcr扩增a)参考引物名称及序列:illumina文库接头引物序列:引物序列来源于试剂盒(nexteradnalibrarypreparationkit,fc-121-1030),i5,i7部分为barcode。nexterap5primer:5’-aatgatacggcgaccaccgagatctacac[i5]tcgtcggcagcgtcnexterap7primer:5’-caagcagaagacggcatacgagat[i7]gtctcgtgggctcggb)将灭菌pcr管置于冰浴中,依次添加各反应组分:成分体积灭菌蒸馏水5.25μl步骤5产物12.5μl5xkapafidelitybuffer5μl10mmeachdntp0.75μlnexterap5primer(10μm)0.5μlnexterap7primer(10μm)0.5μlkapahifidna聚合酶(1u/μl)0.5μl总体积25μlc)使用移液器轻轻吹打10次充分混匀。d)将pcr管置于pcr仪中,设置如下反应程序(hotlid:105℃):7.pcr产物的磁珠纯化利用1×ampurexpbead(beckmancoulter)纯化步骤6得到的pcr产物。a)起始pcr产物体积应为25μl。因pcr过程中样品挥发会导致产物体积不足25μl,进行下面操作之前必须使用灭菌蒸馏水将体积补齐至25μl。b)涡旋震荡混匀ampurexpbeads并吸取25μl体积至含有25μlpcr产物的ep管中,使用移液器轻轻吹打10次充分混匀。室温孵育10分钟。c)步骤b)结束后进行短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心移除上清。d)保持ep管始终处于磁力架中,步骤c)结束后,向ep管中加入200μl新鲜配制的80%(体积百分比)乙醇水溶液漂洗磁珠。室温孵育30秒后小心移除上清。e)重复步骤d),总计漂洗两次。f)保持ep管始终处于磁力架中,开盖,空气中干燥磁珠3分钟。g)将ep管从磁力架中取出,向ep管中加入21μl灭菌超纯水,涡旋振荡或使用移液器轻轻吹打充分混匀,将ep管短暂离心并置于磁力架中分离磁珠和液体。待溶液澄清(约5分钟)小心吸取上清至灭菌ep管中,即得到纯化后的pcr产物,于-20℃保存。8.质量与评估a)质量浓度检测b)利用qubitdsdnahsassaykit(thermofisherscientific)测定,实验组的浓度大于3.5ng/μl。c)电泳检测d)采用1%琼脂糖凝胶检测纯化后的pcr产物。e)agilent2100检测f)将纯化后的pcr产物稀释到合适的浓度,通过bioanalyzerhighsensitivitydnakit(agilent)测定pcr产物片段大小范围。9.使用illumina平台方法进行测序。序列表<110>深圳华大生命科学研究院<120>一种单细胞dna文库的构建方法<160>3<170>siposequencelisting1.0<210>1<211>34<212>dna<213>人工序列()<400>1gtctcgtgggctcggagatgtgtataagagacag34<210>2<211>34<212>dna<213>人工序列()<400>2tacactctttccctacacgacgctcttccgatct34<210>3<211>33<212>dna<213>人工序列()<400>3gatcggaagagcacacgtctgaactccagtcac33当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1