一种纤维素微球载体的制备方法及应用与流程
本发明属于复合材料与生物技术交叉领域,具体涉及一种纤维素微球载体的制备方法及应用。
背景技术:
纤维素是非常充足的可再生、可生物降解的、廉价的生物大分子材料。但是,因为其溶解性问题,纤维素一直得不到大规模的应用。为了提高纤维素的溶解度,目前常用的方法就是使用有机溶剂溶解纤维素,或利用强酸强碱对纤维素进行前处理,将大分子纤维素降解成小分子糖,再加以利用。然而,这些方法中,添加的有机试剂对环境和人体有毒副作用;此外,高温水解处理既高能耗,又高腐蚀,且处理成本高。
技术实现要素:
本发明的目的在于提供一种纤维素微球载体的制备方法及应用,应用高盐溶解微晶纤维素,并通过钙化纤维素凝胶,形成多孔球状载体。其既可以包埋吸附营养物质,作为食品添加剂,生产功能性食品;又可以将包埋农肥,用作缓释肥的载体。
本发明采取的技术方案为:
一种纤维素微球载体的制备方法,包括以下步骤:
a、制备纤维素凝胶;
b、将纤维素凝胶逐滴加入到氯化钙溶液中,振荡反应1~2h,过滤,得到凝胶珠;
c、以无水乙醇清洗凝胶珠;
d、以蒸馏水清洗凝胶珠;
e、室温晾干,即可得到所述纤维素微球载体。
步骤a中,所述纤维素凝胶的制备方法为:将氯化锌溶液倒入微晶纤维素溶液中加热搅拌直至形成均匀澄清的凝胶。如果将微晶纤维素直接加入到氯化锌溶液中或者在加入之后再补加蒸馏水,均会导致微晶纤维素不能完全溶解于氯化锌中。
进一步地,所述氯化锌溶液的浓度为2~3g/ml;所述微晶纤维素溶液的浓度为0.5~1.0g/ml;所述氯化锌溶液与微晶纤维素溶液的体积之比为(5~10):(1~2)。
进一步地,所述加热温度为65℃,搅拌时长30min。
步骤b中,所述滴加的速度为1滴/秒。
步骤b中,所述氯化钙溶液的浓度为18~80g/ml,优选为70g/ml,在此浓度之下对纤维素凝胶进行钙化之后得到的微球载体的硬度较高,且富有弹性,不易破碎。
步骤b中,所述纤维素凝胶与氯化钙溶液的体积之比为(1~2):(4~10)。
步骤c和步骤d中,清洗的次数均为两次,每次清洗30min。凝胶珠需先经无水乙醇清洗再经蒸馏水清洗,这样得到的纤维素微球载体的硬度增加且富有一定的弹性,不易破碎;而如果先以蒸馏水清洗,再以无水乙醇清洗的话,得到的纤维素微球载体的硬度较低且表面较脆,容易破碎。
本发明还提供了根据所述的制备方法制备得到的纤维素微球载体,所述纤维素微球载体为纳米级多孔网状结构,并在纳米级多孔网状结构中均匀分布有微米级骨架结构。
本发明还提供了所述的纤维素微球载体作为食品添加剂、营养物质负载载体、缓释肥料载体的应用。
本发明以安全无毒和高生物相容性的微晶纤维素为基质材料,通过盐溶法制备纤维素凝胶,再通过钙化制备纤维素微球。制成的微球不仅具有优良的吸水保湿性能,还可以作为包埋吸附载体,并具有良好的生物相容性和生物可降解性,在食品加工和农肥等领域具有明显应用价值。本方法反应温和、能耗小、污染少,是一种高效便捷的生产方式。本发明制备的微球,因具有成本低,设备要求不高,且绿色无污染,具有巨大的发展潜力。因此,本发明具有重要的社会意义和应用推广价值。
附图说明
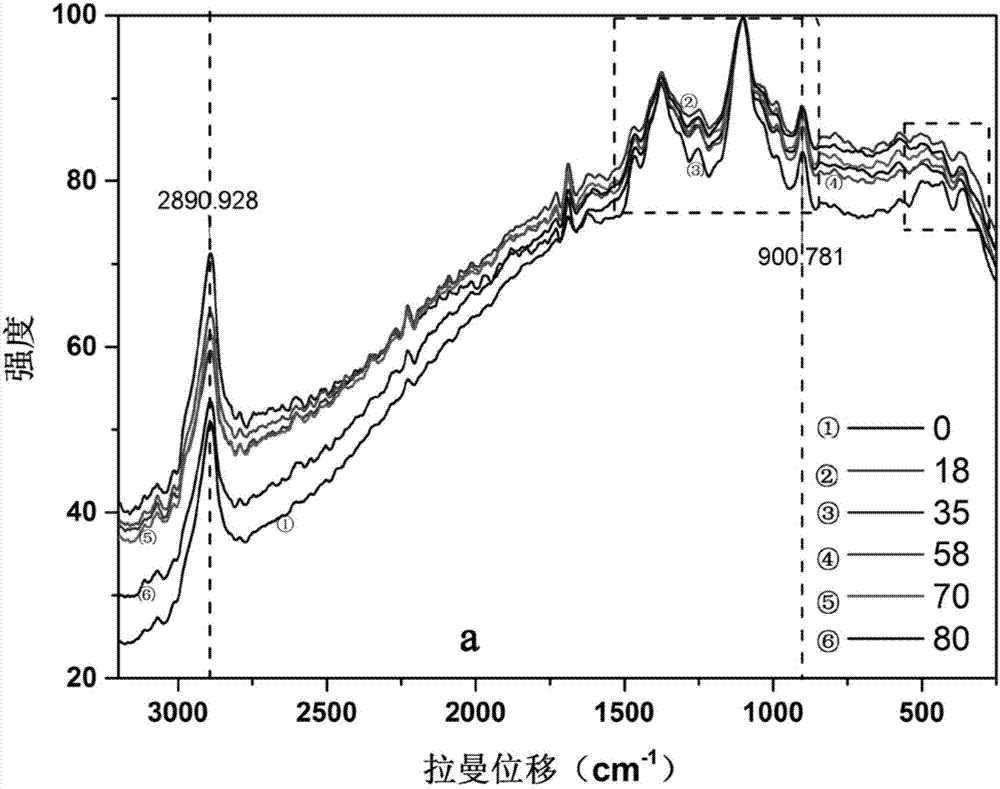
图1为经不同浓度的氯化钙溶液钙化处理的微球的拉曼光谱;
图2为经不同浓度的氯化钙溶液钙化处理的微球的xrd图谱(a);微晶纤维素、微晶纤维素凝胶、氯化锌、氯化钙的xrd图谱(b);
图3为经不同浓度的氯化钙溶液钙化处理的微球的sem图,a-0、b-35%、c-58%、d-70%、e-80%;
图4为经不同处理后纤维素凝胶及其微球的红外图谱(a);微晶纤维素、氯化锌、氯化钙的红外图谱(b);
图5为经不同浓度的氯化钙溶液钙化处理的微球的缓释性;
图6为不同bsa浓度对微球吸附性的影响。
具体实施方式
下面结合实施例及说明书附图对本发明进行详细说明。
本发明中所用的实验材料,如无特殊说明,均为市售;本发明所涉及到的“溶液”,如无特殊说明,均为各物质的水溶液。
实施例1
一种纤维素微球载体的制备方法,包括以下步骤:
a、将10ml3g/ml氯化锌溶液倒入2ml1g/ml微晶纤维素溶液中65℃加热搅拌30min形成均匀澄清的纤维素凝胶;所述氯化锌溶液的制备方法为将氯化锌加入到蒸馏水中65℃加热搅拌溶解30min,直至溶液澄清;
b、将纤维素凝胶逐滴加入到24ml、70g/ml氯化钙溶液中,振荡反应2h,过滤,得到凝胶珠;
c、以无水乙醇清洗凝胶珠2次,每次30min;
d、以蒸馏水清洗凝胶珠2次,每次30min;
e、室温晾干,即可得到所述纤维素微球载体。
实施例2
一种纤维素微球载体的制备方法,包括以下步骤:
a、将5ml2g/ml氯化锌溶液倒入1ml0.5g/ml微晶纤维素溶液中65℃加热搅拌30min形成均匀澄清的纤维素凝胶;所述氯化锌溶液的制备方法为将氯化锌加入到蒸馏水中65℃加热搅拌溶解30min,直至溶液澄清;
b、将纤维素凝胶逐滴加入到12ml、58g/ml氯化钙溶液中,振荡反应1h,过滤,得到凝胶珠;
c、以无水乙醇清洗凝胶珠2次,每次30min;
d、以蒸馏水清洗凝胶珠2次,每次30min;
e、室温晾干,即可得到所述纤维素微球载体。
实施例3
一种纤维素微球载体的制备方法,包括以下步骤:
a、将8ml2.5g/ml氯化锌溶液倒入1.5ml0.8g/ml微晶纤维素溶液中65℃加热搅拌30min形成均匀澄清的纤维素凝胶;所述氯化锌溶液的制备方法为将氯化锌加入到蒸馏水中65℃加热搅拌溶解30min,直至溶液澄清;
b、将纤维素凝胶逐滴加入到19ml、35g/ml氯化钙溶液中,振荡反应1~2h,过滤,得到凝胶珠;
c、以无水乙醇清洗凝胶珠2次,每次30min;
d、以蒸馏水清洗凝胶珠2次,每次30min;
e、室温晾干,即可得到所述纤维素微球载体。
实施例4
其他同实施例1,只是将步骤b中的氯化钙溶液的浓度分别替换为0mg/l、18mg/l、35mg/l、58mg/l、80mg/l,得到的纤维素微球载体的颜色和硬度如表1所示。
表1氯化钙浓度对微球表观特性的影响
注:“+”表示有一定硬度,但稍用力微球仍会破裂;“++”表示硬度增强,并有一定弹性,稍用力微球不会破裂;“+++”表示硬度强,弹性好,用力微球不会破裂
并对得到的各组纤维素微球载体进行raman光谱检测,其raman图谱变化如图1所示。微晶纤维素在1462cm-1和1481cm-1处有两个峰,但当微晶纤维素溶于氯化锌溶液后,这两个峰消失了,表明溶解氯化锌后,再结晶的微晶纤维素的晶体类型可能发生变化。经不同浓度钙化后的微球,其raman图谱总体趋势相同。已知913cm-1附近的峰越宽,其结晶度越高,由表2所示,经钙化后的微球在900cm-1fwhm处的半峰宽大于钙化前的,表明钙化后的微球的结晶度高于未钙化的,尤其是经浓度为58-70%钙离子处理后的微球,结晶度最高。另一方面,1600-150cm-1是纤维素的结晶范围,这个区域的主要的峰位于图1a中左侧的方框内,分别对该区域的峰进行积分,并将其积分面积之和定义为结晶面积(ac)。图1a中右侧方框内的谱图变化较大,将其放大为图1b,由此可见,钙化后的微球在554cm-1和481cm-1有一个偏峰,而没有钙化的微球在这两处没有。因此,554cm-1和481cm-1可能是钙的特征峰位。钙离子浓度为70%时,在481cm-1处的峰最尖,峰值最大。分别对位于481cm-1处的各峰进行积分,其积分面积定义为纤维素-ca晶体面积(aca)。以aca与ac的百分比表示纤维素与钙离子的反应程度,简称钙离子反应度(rdca),rdca越大,表示纤维素与钙离子的反应程度越大。表2显示,钙离子的反应程度与纤维素-ca的结晶度几乎呈正相关,钙离子反应程度越高,复合物的结晶度越高,其中,当钙离子浓度为70%时,形成纤维素-钙复合物的程度最高。
表2不同钙化处理的微球的拉曼特征光谱分析
注:rdca=100*aca/ac
对得到的各组纤维素微球载体进行xrd检测,如图2所示,微晶纤维素主要有三个主峰及若干偏峰,如图2b所示,对应的晶向分别为101、021、002和040面。微晶纤维素溶于氯化锌溶液后,其主要晶向及其晶体比例发生明显变化,表明氯化锌高盐溶液改变了微晶纤维素原来的晶体形成走向,势必会进一步影响纤维素-zn的晶体结构。钙化后的纤维素三元复合物(纤维素-zn-ca)的晶向更复杂,表明钙化导致晶体形成呈现多向性,晶体尺寸变化范围扩大。其中,不同浓度钙处理后的微球晶向大致相同,但不同尺寸的晶体比例有变化。综上所述,高盐溶解和钙化过程能显著改变纤维素的晶向及晶体大小,进而一定程度影响了纤维素微球的微观结构及其强度。
经sem检测后,其微观结构变化如图3所示,微球基质呈纳米级多孔网状结构,随着钙离子浓度的增加,多孔基质中出现微米级骨架结构,并均匀分布在多孔网状结构中;同时,其多孔网状结构也随着钙离子浓度的增加,而越显规则,尤其是在钙离子浓度为70%时,多孔结构呈现规整的层状结构。由此表明,随着钙离子浓度的增加,微球的机械强度应会相应增加。
实施例5
为分析纤维素微球过程中钙化、水洗和醇洗过程的作用机理,分别制备纤维素凝胶(a)、经水洗的纤维素凝胶(b)、经醇洗的纤维素凝胶(c)和水洗后的钙化纤维素微球(d)四个样本。
纤维素凝胶(a)的制备方法同实施例1中的步骤a。
水洗的纤维素凝胶(b)的制备方法为:将钙化前纤维素凝胶(a)转移到蒸馏水中清洗30分钟,再重复清洗一次。凝胶迅速收缩,表面开始硬化,但硬化后的凝胶表面非常脆弱,易破碎。
经醇洗的纤维素凝胶(c)的制备方法为:将钙化前纤维素凝胶(a)转移到无水乙醇清洗30分钟,再重复清洗一次。凝胶迅速收缩,表面开始硬化,硬化后的凝胶表面具有一定弹性,不易破碎。
水洗后的钙化纤维素微球(d)的制备方法为:将实施例1中的步骤b钙化形成的纤维素微球先转移到无水乙醇中清洗30分钟,再重复清洗一次;微球缩小,变硬且具有一定的弹性。再将微球转移到蒸馏水中清洗30分钟,再重复清洗一次。微球硬度增加,用力按压,富有弹性,不易破碎。
并对这四个样本进行红外检测,由图1所示:与纤维素粉末的红外图谱比较,纤维素溶解于氯化锌溶液后,其在3332cm-1出的峰位移至3394cm-1,且半峰高增大,表明氢键类型发生变化(即分子间氢键可能向分子内氢键转变),且振动增强,3200cm-1处出现一个偏锋。3455–3410cm-1和3375–3340cm-1分别表示o2h···o6和o3h···o5的分子内氢键,3310–3230cm-1表示o6h···o3的分子间氢键。由此表明纤维素溶解于氯化锌溶液后,纤维素分子间氢键增加,分子内氢键类型发生变化,表明纤维素分子中的氢键发生了断裂与重新缔合的重排过程。1619cm-1处出现一个新的强峰(为锌离子特征峰),表明凝胶中含有大量的锌离子,由此推断,凝胶中的锌离子可能与纤维素分子中的羟基发生反应。1068cm-1处和1022cm-1的峰值显著降低,1754cm-1、1654cm-1、1569cm-1和136cm-19处的峰消失,这可能是因为纤维素分子与锌离子的作用导致的。
水洗过程改变了纤维素凝胶的红外图谱:水洗后的纤维素-zn凝胶的红外图谱更接近与原始的纤维素粉末。相对于水洗前,1619cm-1峰值的锌离子特征峰值明显降低,表明水洗将凝胶内多余的自由锌离子洗脱,导致锌离子含量降低。原本位于3394cm-1处的宽峰回复位移至3347cm-1,但其峰强高于纤维素粉末对应的峰强,表明氢键数目相对增加,由此推断,该凝胶内部由于锌离子的进入导致其纤维分子间增大,水分子充分进入,进而形成大量的氢键。1068cm-1处和1022cm-1的峰值相对于水洗前,显著增加,可能是由于大量锌离子洗脱所致。
醇洗后的纤维素-zn凝胶红外图谱与纤维素-zn凝胶的红外图谱相比,其在2981cm-1、2919cm-1和875cm-1处各出现了一个新峰,可能是乙醇洗涤多余锌离子时,进入纤维素分子内,其羟基与纤维素分子羟基形成氢键以及乙醇-ch振动的结果。正由于氢键的形成,将乙醇分子的c-c骨架引入到纤维素分子间,这可能就是纤维素-zn凝胶经醇洗后硬度明显增强的缘故。1619cm-1锌离子特征峰减弱,1022cm-1处的峰值显著增加,表明乙醇洗涤过程清除了多余的锌离子。相对于纤维素-zn凝胶的红外图谱,醇洗导致3200cm-1处出现一个偏锋强度减弱,表明醇洗过程一定程度上也改变了纤维分子中的氢键类型。与纤维素粉末、纤维素-zn凝胶和水洗后的纤维素-zn凝胶比较,醇洗后的纤维素-zn凝胶4000-3000cm-1区段红外图谱,与纤维素-zn更相似,表明醇洗的过程并没有像水洗过程那样显著改变纤维分子中的氢键类型。
3494cm-1和3440cm-1是氯化钙的特征峰,钙化后的微球在这两处出现了微弱的峰,表明钙化后的微球含有钙离子。水洗后的钙化纤维素微球,4000-3000cm-1区段的宽峰峰值位于3347cm-1处,这可能是水洗的作用所致。1072cm-1和1010cm-1相对于3200-3400cm-1处的比值显著增强,可能是由于钙化过程中钙离子置换了纤维素-zn凝胶中的锌离子。1619cm-1处的强峰位移至1639cm-1处,并且半峰高降低。1454cm-1、1369cm-1和1311cm-1处出现新峰。可能是锌离子被钙离子置换所致。
实施例6
纤维素微球载体的保水性测试
将新制备的微球颗粒,经无水乙醇反复清洗两次,每次清洗30min,再经不同时间的蒸馏水清洗后,发现其保水性差异明显,具体结果如表3所示:
表3不同清洗时间对微球保水性的影响
附:“+++”表示微球表面吸附有明显的水珠,保水性优良;“++”表示微球表面吸附有水珠,保水性良好;“+”表示微球表面附有少量水分,具有一定保水性;“-”表示微球表面失水干燥;“--”表示微球内部失水,质量减少。
由此表明:随着微球洗涤时间的延长,导致微球内部盐离子不断流失,致使微球锁水能力降低。因此,可以通过降低微球洗涤的时间,提升其保水性。这样形成的微球载体,在使用过程中,不仅可以达到保水保湿的效果,而且可以不断向外环境缓释zn2+和ca2+,以达到补锌补钙的目的。
实施例7
纤维素微球载体的缓释性测试
将50mg经不同浓度氯化钙钙化后的微球,浸泡在20ml蒸馏水溶液中,经过多轮换水后,至微球质量不在变化为止,室温干燥后,称量微球的质量。结果如图5所示,随着钙化时使用氯化钙溶液浓度的增加,其形成的微球在前期洗涤的基础上,再经充分洗涤后发现,其质量流失占其总质量的百分比逐渐增加,当钙浓度达到70%时,质量流失达到最高值,约流失39%。表明钙离子70%时,微球的充分洗涤需要更长的时间,由此推断形成的微球结构较为致密,质量流失较缓慢。由此推断,其形成的微球具有较佳的缓释功能。
实施例8
纤维素微球载体的蛋白吸附性测试
分别称取几份0.3g干燥的微球,放入浓度为0.5-3.0mg/ml牛血清蛋白醋酸缓冲溶液中,40h后,检测残留蛋白的浓度,计算吸附量。结果如图6所示,随着bsa浓度的增加,微球的吸附量持续增加,进一步分析发现,随着bsa浓度的增加,微球的吸附效率显著提升,最高超过原始吸附效率的105%,表明纤维素微球具有较好的吸附性能。
吸附效率增加值=100*(m-m0)/(n-n0)-100,m和n分别表示待测样本的微球吸附量和bsa浓度,m0和n0分别表示bsa浓度为0.5mg/ml时的微球吸附量和bsa浓度。
比较例1
对纤维素凝胶溶液配制时,原料的加入方式的测试
(1)称取0.5-1g微晶纤维素粉末,直接倒入浓度为2-3g/ml氯化锌溶液中,搅拌均匀后,静置于65℃水浴中30分钟。纤维素悬浮于氯化锌溶液中,呈乳白色浑浊状,表明纤维素不溶于氯化锌溶液。
(2)称取0.5-1g微晶纤维素粉末,直接倒入浓度为2-3g/ml氯化锌溶液中,再加入1-2ml蒸馏水,搅拌均匀后,静置于65℃水浴中30分钟。溶液呈浑浊的胶体状,表明纤维素不完全溶解于氯化锌溶液中。
(3)向0.5-1g微晶纤维素粉末加入1-2ml蒸馏水,搅拌均匀后;将浓度为2-3g/ml氯化锌溶液倒入纤维素乳浊液中,再搅拌均匀后,静置于65℃水浴中30分钟。溶液呈透明的胶体状,表明纤维素完全溶解于氯化锌溶液中。
上述参照实施例对一种纤维素微球载体的制备方法及应用进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!