一种左乙拉西坦的合成工艺的制作方法
本发明涉及医药制备技术领域,特别涉及一种抗癫痫药物的制备方法。
背景技术
癫痫是一种短暂性大脑功能性失常的慢性综合征,由神经元突触病变引起的常见疾病。据了解,全球范围内癫痫患者达数千万之多,少数人可完全治疗,多数人需要坚持长期使用抗癫痫药物,以减少病情发作,为患者创造良好的生活品质。经历了百年发展,从最初使用的溴化钾到苯巴比妥的出现,再到上世纪年代,苯妥英、丙戊酸等的问世,治疗癫痫领域有了很大进展。左乙拉西坦为吡咯烷酮类抗癫痫药物,可有效控制癫痫发作,具有治疗指数高、安全性好、副作用轻微等特点,是一款具有预防作用的广谱抗癫痫药物,其化学结构式如下式所示:
1987年,比利时ucb公司公开了一种采用化学拆分法合成左乙拉西坦的方法。以手性苯乙胺为拆分试剂,苯为溶剂,拆分得到(s)-左乙拉西坦酸;再通过氯甲酸乙酯制得混合酸酐,最后氨解获得左乙拉西坦。该工艺路线中采用苯作为溶剂(一类溶剂),显然不符合当前ichq3指南的要求。同时,工艺中采用的氯甲酸乙酯,因其高毒性,是被列入管制品清单的化合物。虽然,早期的工艺已不符合时代发展的新要求,但是它对新工艺开发和优化具有非常重要的指导意义。具体合成工艺如下式所示:
2005年,美国专利us2005/0182262公开了一种以(s)-2-氨基丁酸盐酸盐为起始原料,通过酯化和氨解反应,制得(s)-2-氨基丁酰胺盐酸;再与4-氯丁酰氯反应,得到(s)-2-(4-氯丁酰胺)丁酰胺;最后环合得到左乙拉西坦。该路线在以(s)-2-氨基丁酸为起始原料,避免了拆分得到手性化合物的操作。但是在酯化过程中,需要用到氯化亚砜、草酰氯或五氯化磷等高毒性、强腐蚀性的试剂,无论是对反应设备或人员操作要求都比较高,同时反应会产生大量的酸性废液,环保压力大。具体合成工艺如下式所示:
2013年,中国专利cn102558012报道了一种新的合成左乙拉西坦方法,以(s)-2-氨基丁酸为原料与4-氯丁酰氯进行酰化反应制得(s)-2-(4-氯丁酰胺)丁酸,再与氯甲酸乙酯等酰化剂反应得到混合酸酐,然后通过氨解制得(s)-2-(4-氯丁酰胺)丁酰胺;最后在相转移催化剂的存在下进行环合反应得到左乙拉西坦。该路线采用廉价易得的(s)-2-氨基丁酸为起始原料,同样避免了拆分的操作。但是在从羧酸转化为酰胺的过程中,采用了氯甲酸烷基酯类剧毒管制品。在商业化大生产中,存在很大的安全隐患和安全方面的成本投入。同时,该工艺所得的左乙拉西坦成品,光学异构体偏高。具体合成工艺如下式所示:
技术实现要素:
本发明的目的在于克服现有技术的不足,提供一种无须化学拆分,不使用剧毒或强腐蚀性的化学试剂,操作简单、条件温和、环境友好和成品质量高的的左乙拉西坦合成工艺。
为了达到上述目的,本发明所设计的一种左乙拉西坦的合成工艺,合成路线为:
它包括如下具体步骤:
第一步:(s)-2-(4-氯丁酰胺)丁酰胺的合成
在反应瓶中加入溶剂,搅拌下加入(s)-2-(4-氯丁酰胺)丁酸,搅拌溶解后加入(boc)2o,控制反应体系内温度为20~30℃,缓慢滴加吡啶至反应体系中,滴加时间为20分钟,加完毕后搅拌反应30分钟;反应体系中加入铵盐,在20~30℃下,搅拌反应5~8小时;反应完毕后,过滤除去体系中的不溶物,滤液减压浓缩至干;浓缩物加到丙酮中,加热到60~70℃溶解至全部溶清,冷却至10~20℃搅拌析晶2~3小时;过滤、减压干燥得到(s)-2-(4-氯丁酰胺)丁酰胺;
第二步:左乙拉西坦的合成
在反应瓶中加入溶剂,搅拌下加入(s)-2-(4-氯丁酰胺)丁酰胺,搅拌溶清之后将体系冷却至0~5℃;分批加入碱,0~5℃下继续搅拌3~4小时;反应完毕后,用4n盐酸调节体系的ph值为6~7;减压浓缩除去溶剂后,加入二氯甲烷和纯化水,分液萃取,水相再用二氯甲烷萃取;合并有机相,用无水硫酸钠干燥,过滤浓缩,所得粗品用精制溶剂重结晶得到左乙拉西坦纯品。
作为优选,第一步所述溶剂选自醇类、醚类、烃类、酮类、酯类中的一种或几种,优选的可以是:乙腈、二氯甲烷、三氯甲烷、二氧六环、dmf等。
作为优选,第一步所述(boc)2o与(s)-2-(4-氯丁酰胺)丁酸的摩尔比为0.3~2.0,特别优选为0.5~1.5,尤其是1.2。
作为优选,第一步所述吡啶与(s)-2-(4-氯丁酰胺)丁酸的摩尔比为0.1~1.5,特别优选为0.5~1.0,尤其是0.8。
作为优选,第一步所述铵盐的种类为碳酸氢铵、碳酸铵、醋酸铵、甲酸铵、氟化铵、氯化铵、溴化铵、碘化铵中的一种或多种。
作为优选,第一步所述铵盐与(s)-2-(4-氯丁酰胺)丁酸的摩尔比为0.1~3.0,特别优选为0.5~2.0,尤其是1.2。
作为优选,第二步所述溶剂选自醇类、醚类、烃类、酮类、酯类中的一种或几种,优选的可以是:乙腈、二氯甲烷、四氢呋喃、二氧六环、dmf和dmso等。
作为优选,第二步所述碱选自有机碱或者无机碱,优选的可以是:甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾、lda、lihmds、nahmds,正丁基锂、异丁基锂、叔丁基锂等。
作为优选,第二步所述碱与(s)-2-(4-氯丁酰胺)丁酰胺的摩尔比为0.5~4.5,特别优选为1.5~3.0,尤其是2.5。
作为优选,第二步所述的左乙拉西坦粗品精制溶剂选自:乙酸乙酯、甲酸乙酯、乙酸正丁酯、甲基叔丁基醚、乙醚、异丙醚、四氢呋喃、丙酮、甲基乙基酮中的一种或多种。
本发明针对现有技术合成左乙拉西坦工艺中存在的不足:1、化学拆分过程中存在收率低(最高理论产率50%)、使用不符合ichq3要求的溶剂(苯);2、从羧酸中间体制备酰胺中间体时,需要使用剧毒或强腐蚀性的化学试剂,例如:氯甲酸乙酯、氯甲酸甲酯、氯化亚砜、五氯化磷等;3、左乙拉西坦成品光学异构体偏高等;开发了一条操作简单、条件温和、环境友好和成品质量高的左乙拉西坦工艺路线,即以(s)-2-(4-氯丁酰胺)丁酸为起始原料,以吡啶作为碱、(boc)2o为羧酸的活化试剂,加入铵盐,制得了(s)-2-(4-氯丁酰胺)丁酰胺,最后在碱存在下进行环合反应得到左乙拉西坦。
本发明最大的特点在于:1、避免了拆分得到手性化合物的操作,同时光学纯度高,成品质量高;不采用苯作为溶剂(一类溶剂),符合当前ichq3指南的要求;不需要用到强腐蚀性的试剂,对反应设备或人员操作要求不高,同时反应不会产生大量的酸性废液,环保压力小;2、不需要用到氯化亚砜、草酰氯、五氯化磷、氯甲酸乙酯、氯甲酸烷基酯类等高毒性试剂,在商业化大生产中,安全隐患小,安全投入成本低;3、反应条件温和,后处理简单,经济环保,适合工业化生产。
附图说明
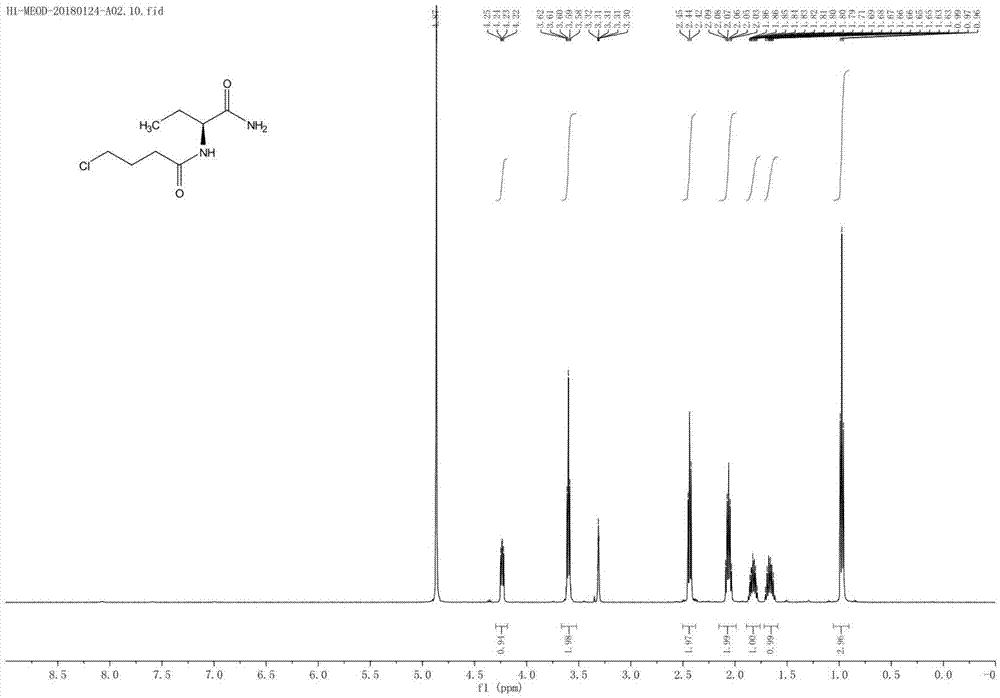
图1为本发明中(s)-2-(4-氯丁酰胺)丁酰胺的核磁共振谱图。
图2为本发明得到的左乙拉西坦的核磁共振谱图。
具体实施方式
实施例1:
第一步:(s)-2-(4-氯丁酰胺)丁酰胺的合成
2l反应瓶中,加入乙腈1000ml,搅拌下加入(s)-2-(4-氯丁酰胺)丁酸(100.0g,481.6mmol),搅拌溶解后,加入(boc)2o(126.1g,577.8mmol),控制反应体系内温度为20~30℃,缓慢滴加吡啶(26.7g,337.6mmol)至反应体系,滴加时间为20分钟,加完毕后搅拌反应30分钟;加入碳酸氢铵(45.7g,578.1mmol),在20~30℃下,搅拌反应5~8小时;tlc点板检测原料反应完毕(展开剂:乙酸乙酯:冰醋酸=100:1),过滤除去体系中的不溶物,滤液减压浓缩至干;浓缩物加到丙酮中,加热到60~70℃溶解至全部溶清,冷却至10~20℃搅拌析晶2~3小时;过滤、减压干燥得到(s)-2-(4-氯丁酰胺)丁酰胺81.2克,收率81.6%,纯度98.2%。
第二步:左乙拉西坦的合成
1l反应瓶中,加入四氢呋喃600ml,搅拌下加入(s)-2-(4-氯丁酰胺)丁酰胺(50.0g,241.9mmol),搅拌溶清之后,将体系冷却至0~5℃;分批加入叔丁醇钾(67.9g,605.1mmol),0~5℃下继续搅拌3~4小时;tlc点板检测原料反应完毕(展开剂:乙酸乙酯:冰醋酸=100:1),用4n盐酸调节体系的ph值为6~7;减压浓缩除去四氢呋喃后,加入二氯甲烷500ml和纯化水150ml,分液萃取,水相再用二氯甲烷200ml萃取;合并有机相,用无水硫酸钠干燥,过滤浓缩;所得粗品用混合溶剂(乙酸乙酯/甲基叔丁基醚)重结晶得到左乙拉西坦纯品32.5克,收率78.9%,光学纯度99.9%。
实施例2:
第一步:(s)-2-(4-氯丁酰胺)丁酰胺的合成
2l反应瓶中,加入乙腈1000ml,搅拌下加入(s)-2-(4-氯丁酰胺)丁酸(100.0g,481.6mmol),搅拌溶解后,加入(boc)2o(126.1g,577.8mmol),控制反应体系内温度为20~30℃,缓慢滴加吡啶(26.7g,337.6mmol)至反应体系,滴加时间为20分钟,加完毕后搅拌反应30分钟;加入碳酸铵(37.0g,385.3mmol),在20~30℃下,搅拌反应5~8小时;tlc点板检测原料反应完毕(展开剂:乙酸乙酯:冰醋酸=100:1),过滤除去体系中的不溶物,滤液减压浓缩至干;浓缩物加到丙酮中,加热到60~70℃溶解至全部溶清,冷却至10~20℃搅拌析晶2~3小时;过滤、减压干燥得到(s)-2-(4-氯丁酰胺)丁酰胺76.0克,收率76.4%,纯度97.1%。
第二步:左乙拉西坦的合成
1l反应瓶中,加入四氢呋喃100ml,搅拌下加入(s)-2-(4-氯丁酰胺)丁酰胺(50.0g,241.9mmol),搅拌溶清之后,将体系冷却至0~5℃;分批加入lihmds(1mol/linthf,605.0ml,605.0mmol),0~5℃下继续搅拌3~4小时;tlc点板检测原料反应完毕(展开剂:乙酸乙酯:冰醋酸=100:1),用4n盐酸调节体系的ph值为6~7;减压浓缩除去四氢呋喃后,加入二氯甲烷500ml和纯化水150ml,分液萃取,水相再用二氯甲烷200ml萃取;合并有机相,用无水硫酸钠干燥,过滤浓缩;所得粗品用混合溶剂(乙酸乙酯/异丙醚)重结晶得到左乙拉西坦纯品29.6克,收率71.9%,光学纯度99.9%。
- 还没有人留言评论。精彩留言会获得点赞!