一种合成α-胺基酮衍生物的方法与流程
本发明涉及一种α-胺基酮衍生物的合成方法,尤其涉及一种使用pd催化羧酸与2h-氮杂环丙烷亲核加成、开环重排反应合成α-胺基酮衍生物的新方法,属于有机合成技术领域。
背景技术
α-胺基酮衍生物是非常重要的结构单元,在药物和天然产物领域应用广泛。作为重要的合成中间体,α-胺基酮可以用于合成多种杂环化合物,例如咪唑、噁唑、噻唑及其他的精细化学品。目前报道的合成α-胺基酮衍生物的方法包括:cope重排、neber重排、分子间醛-亚胺交叉偶联反应、dakin-west等反应,但是这些方法往往存在以下问题:起始原料制备复杂、反应条件苛刻、底物范围受限。由此可见,发展新的合成α-胺基酮衍生物的方法具有重要的意义。
2h-氮杂环丙烷环张力大,反应活性高,主要通过三种断裂方式发生开环反应:1)c-n键断裂生成烯基乃春,2)过渡金属催化或者光催化c-c键断裂生成腈偶极子,3)c-c和c-n键同时断裂生成卡宾中间体。然而,2h-氮杂环丙烷与羧酸经由亲核加成反应、分子内开环重排串联反应还未见报道。
根据文献【yadavj.s.,reddyb.v.s.,sadashivk.,etal.tetrahedronlett.,2002,43,2099-2101;lix.,lig.,changh.h.,etal.rscadv.,2014,4,6490–6495.】所述,氮杂环丙烷与羧酸发生开环反应,合成路线如下:
该方法利用羧酸中羧基上富电子氧的亲核性质,完成了羧酸对氮杂环丙烷的开环反应。此方法使用的氮杂环丙烷必须使用对甲苯磺酰基进行保护,鉴于其张力作用,具备开环能力,但只能经历亲核开环反应,反应类型单一。此反应得到的产物为对甲苯磺酰基保护的氨基化合物,反应的区域选择性差。
根据文献【oh,b.;nakamura,i.;yamamoto,y.j.org.chem.2004,69,2856-2858】所述,亚甲基氮杂环丁烷与羧酸发生开环反应,合成路线如下:
该方法虽说能制备α-胺基酮衍生物,但使用的亚甲基氮杂环丁烷制备复杂,底物的范围很窄,并且只能得到叔酰胺产物。鉴于膦配体的存在,反应需要严格的无氧条件,限制了其应用。
技术实现要素:
本发明的主要目的在于提供一种合成α-胺基酮衍生物的方法,以克服现有技术的不足。
本发明实施例提供了一类2h-氮杂环丙烷作为合成子在合成α-胺基酮衍生物中的应用。
优选的,所述2h-氮杂环丙烷的结构式为
本发明实施例还提供了一类羧酸作为合成子在合成α-胺基酮衍生物中的应用。
本发明实施例还提供了一种合成α-胺基酮衍生物的方法,其包括:使包含2h-氮杂环丙烷、羧酸类化合物、催化剂、添加剂、碱和溶剂的混合反应体系反应,获得α-胺基酮衍生物;
其中,所述2h-氮杂环丙烷的结构式为
在一些实施例中,所述方法具体包括:将2h-氮杂环丙烷、羧酸类化合物、催化剂、添加剂、碱和溶剂混合均匀,形成混合反应体系,并于80~110℃反应12~18h,经历亲核加成反应、开环重排反应,获得一系列α-胺基酮衍生物。
本发明实施例还提供了由前述方法制备的α-胺基酮衍生物。
进一步地,所述α-胺基酮衍生物具有α-胺基酮结构单元,其结构式为
较之现有技术,本发明的有益效果在于:
1)本发明提供的合成α-胺基酮衍生物的方法首次使用2h-氮杂环丙烷作为合成子,经由亲核加成反应、开环重排反应,成功实现一步构建α-胺基酮衍生物的方法。因此,本发明实现了2h-氮杂环丙烷与羧酸的亲核加成/开环串联反应,高产率地制备出多种多取代的α-胺基酮衍生物;
2)本发明提供的合成α-胺基酮衍生物的方法首次将2h-氮杂环丙烷合成子用于亲核加成反应、开环重排反应。该方法使用简单易得的羧酸作为合成子,经历亲核加成、开环重排反应,为扩充2h-氮杂环丙烷的反应类型提供了支持;
3)本发明提供的合成α-胺基酮衍生物的方法选择的催化剂体系同时满足亲核加成反应、开环重排反应的需求,大大提高了反应的原子利用率,简化反应步骤,充分体现了汇聚合成方法的优势;
4)本发明提供的合成α-胺基酮衍生物的方法对水和空气稳定不敏感,反应无需惰性气体保护,无需在惰性气体中进行,操作简单,反应条件温和,反应效率高,底物的适用范围广,原子利用率高,反应区域选择性和化学选择性高,多种官能团都适用于此反应条件。对于杂环类底物,该方法也能取得很好的反应效果;
5)本发明制备的α-胺基酮衍生物结构新颖,具有潜在的应用价值。
附图说明
图1是本发明一典型实施例中合成α-胺基酮衍生物的方法的化学反应原理示意图。
图2是本发明实施例1所获α-胺基酮衍生物3aa的核磁共振图谱之氢谱。
图3是本发明实施例1所获α-胺基酮衍生物3aa的核磁共振图谱之碳谱。
图4是本发明实施例12所获α-胺基酮衍生物3ba的核磁共振图谱之氢谱。
图5是本发明实施例12所获α-胺基酮衍生物3ba的核磁共振图谱之碳谱。
图6是本发明实施例13所获α-胺基酮衍生物3ca的核磁共振图谱之氢谱。
图7是本发明实施例13所获α-胺基酮衍生物3ca的核磁共振图谱之碳谱。
图8是本发明实施例14所获α-胺基酮衍生物3da的核磁共振图谱之氢谱。
图9是本发明实施例14所获α-胺基酮衍生物3da的核磁共振图谱之碳谱。
图10是本发明实施例1所获α-胺基酮衍生物3aa的高分辨质谱图。
图11是本发明实施例12所获α-胺基酮衍生物3ba的高分辨质谱图。
图12是本发明实施例13所获α-胺基酮衍生物3ca的高分辨质谱图。
图13是本发明实施例14所获α-胺基酮衍生物3da的高分辨质谱图。
具体实施方式
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案,主要是一类pd催化亲核加成、开环重排串联反应,合成α-胺基酮衍生物的新方法,实现亲核加成、开环重排串联反应,一步构建具有多种官能团的α-胺基酮衍生物。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
2h-氮杂环丙烷往往会与其他的不饱和键发生环化反应,进而引发开环反应,而能与2h-氮杂环丙烷中的亚胺发生亲核加成反应/开环重排反应的的试剂很少。该方法是用简单易得的羧酸类化合物作为合成子,经历亲核加成、开环重排反应,为扩充2h-氮杂环丙烷的反应类型提供了支持。
与氮杂环丙烷不同,2h-氮杂环丙烷既有三元环的张力,还包含一个亚胺基团,反应活性更高,反应类型也更加丰富;与亚甲基氮杂环丁烷相比,2h-氮杂环丙烷制备方法简单。因此通过2h-氮杂环丙烷与羧酸的亲核加成、开环串联反应构建α-胺基酮衍生物的研究备受期待。
本发明实施例的一个方面提供了一类2h-氮杂环丙烷作为合成子在亲核加成、开环重排串联反应中的应用。
本发明实施例的另一个方面提供了一类2h-氮杂环丙烷作为合成子在合成α-胺基酮衍生物中的应用。
进一步地,所述2h-氮杂环丙烷的结构式为
具体的,所述r1包括芳基(取代基团包括:氢原子、甲基、乙基、cl-、f-、br-、i-、cf3-或-ome等,但不限于此)或者杂芳基(包含噻吩、呋喃、吡啶等),所述r2包括芳基(取代基团包括:氢原子、甲基、乙基、cl-、f-、br-、i-、cf3-、co2et-或-ome等,但不限于此)或者杂芳基(包含噻吩、呋喃、吡啶等)。
在一些实施例中,所述应用包括:采用2h-氮杂环丙烷作为合成子在亲核加成、开环重排串联反应合成α-胺基酮衍生物中的应用。
本发明实施例的另一个方面还提供了一类羧酸类化合物在合成α-胺基酮衍生物中的应用。
进一步地,所述羧酸类化合物的结构式为
具体的,所述r3中所述芳基取代基包括甲基、乙基、cl-、f-、br-、i-、cf3-、meo-、苯基或co2et-等,但不限于此;所述杂芳基包括噻吩、呋喃、吡啶等,但不局限于此;所述烷基包括甲基、乙基、丙基、-cf3等,但不局限于此。
本发明实施例的另一个方面还提供了一种合成α-胺基酮衍生物的方法,其包括:使包含2h-氮杂环丙烷、羧酸类化合物、催化剂、添加剂、碱和溶剂的混合反应体系反应,获得α-胺基酮衍生物。
其中,所述2h-氮杂环丙烷的结构式为
其中,所述羧酸类化合物的结构式为
在一些实施例中,如图1所示,所述方法具体包括:将2h-氮杂环丙烷、羧酸类化合物、催化剂、添加剂、碱和溶剂混合均匀,形成混合反应体系,并于80~110℃反应12~18h,经历亲核加成反应、开环重排串联反应,获得一系列α-胺基酮衍生物。
在一些实施例中,所述2h-氮杂环丙烷与催化剂的摩尔比为10~100:1。
在一些实施例中,所述羧酸类化合物与催化剂的摩尔比为10~100:1。
在一些实施例中,所述2h-氮杂环丙烷与羧酸类化合物的摩尔比为0.8~50:1。
在一些实施例中,所述添加剂与羧酸类化合物的摩尔比为0.1~1:1。
在一些实施例中,所述碱与羧酸类化合物的摩尔比为0.1~1:1。
在一些实施例中,在所述混合反应体系中,所述羧酸类化合物或2h-氮杂环丙烷的浓度为0.1~1mol/l。
进一步地,所述催化剂包括商业易得的pd(oac)2,但不限于此。
进一步地,所述添加剂包括银盐添加剂和dmap(4-二甲氨基吡啶)等添加剂,但不限于此。
进一步地,所述银盐添加剂包括氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银,但不限于此。
进一步地,所述碱包括li2co3,但不限于此。
进一步地,所述溶剂范围较广,所述溶剂包括醚类溶剂、醇类溶剂、甲苯、酰胺类溶剂和乙腈中的任意一种或两种以上的组合,但不限于此。
更进一步地,所述醚类溶剂包括1,4-二氧六环,但不限于此。
更进一步地,所述酰胺类溶剂包括n,n-二甲基甲酰胺,但不限于此。
更进一步地,所述醇类溶剂包括乙醇,但不限于此。
优选的,所述α-胺基酮衍生物包括多种官能团取代的衍生物,例如:me-,f-,cf3-,meo-,cl-,br-,co2et-等。
例如,在一些优选实施案例之中,所述方法的化学反应式可以是:
其中,r1包括芳基(取代基团包括:氢原子、甲基、乙基、cl-、f-、br-、i-、cf3-或-ome等,但不限于此)或者杂芳基(包含噻吩、呋喃、吡啶等);所述r2包括芳基(取代基团包括:氢原子、甲基、乙基、cl-、f-、br-、i-、cf3-、co2et-或-ome等,但不限于此)或者杂芳基(包含噻吩、呋喃、吡啶等)。所述r3包括芳基、杂芳基或者烷基,所述芳基取代基包括甲基、乙基、cl-、f-、br-、i-、cf3-、meo-、苯基或co2et-等,但不限于此;所述杂芳基包括噻吩、呋喃、吡啶等,但不局限于此;所述烷基包括甲基、乙基、丙基、-cf3等,但不局限于此。
在一些实施例中,所述方法还包括:反应结束后,将所述混合反应体系冷却至室温,除去溶剂,分离,获得α-胺基酮衍生物。
进一步地,所述方法的产率为19~92%。
其中,在一更为优选的实施方案之中,本发明的方法可以具体包括如下步骤:
于反应容器内加入pd(oac)2、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银、dmap、li2co3、2h-氮杂环丙烷2、羧酸类化合物1、溶剂,升温至80~110℃,反应12~18h。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3。
本发明首次使用2h-氮杂环丙烷类化合物作为合成子用于亲核加成、开环重排串联反应。2h-氮杂环丙烷类化合物易在金属催化剂、光催化的条件下发生c-c或者c-n键断裂,再与不饱和键发生[3+n]环加成反应,而该方法采用2h-氮杂环丙烷类化合物作为合成子,经历亲核加成、开环重排反应,为扩充2h-氮杂环丙烷化合物的反应类型提供了支持。
另外,该方法经历串联过程,经由亲核加成反应、开环重排反应,是高效地汇聚合成方法。而且,本发明反应的底物适用范围广,多种官能团都适用于此反应条件。对于多种杂环底物,该方法也能取得很好的反应效果。
本发明实施例的另一个方面还提供了由前述方法制备的α-胺基酮衍生物。
进一步地,所述α-胺基酮衍生物具有α-胺基酮结构单元,其结构式为
优选的,所述α-胺基酮衍生物包括多种官能团取代的衍生物,例如:me-,f-,cf3-,meo-,cl-,br-,co2et-等,但不限于此。
藉由上述技术方案,本发明首次使用2h-氮杂环丙烷类化合物作为合成子,经由亲核加成反应、开环重排串联反应,成功一步构建α-胺基酮衍生物。本发明对水和空气稳定不敏感,操作简单,反应条件温和,反应效率高,底物的适用范围广,原子利用率高,反应区域选择性和化学选择性高,所制备的α-胺基酮衍生物结构新颖,具有潜在的应用价值。
下面结合若干优选实施例及附图对本发明的技术方案做进一步详细说明,但本发明并不仅仅局限于下述实施例。
实施例1
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
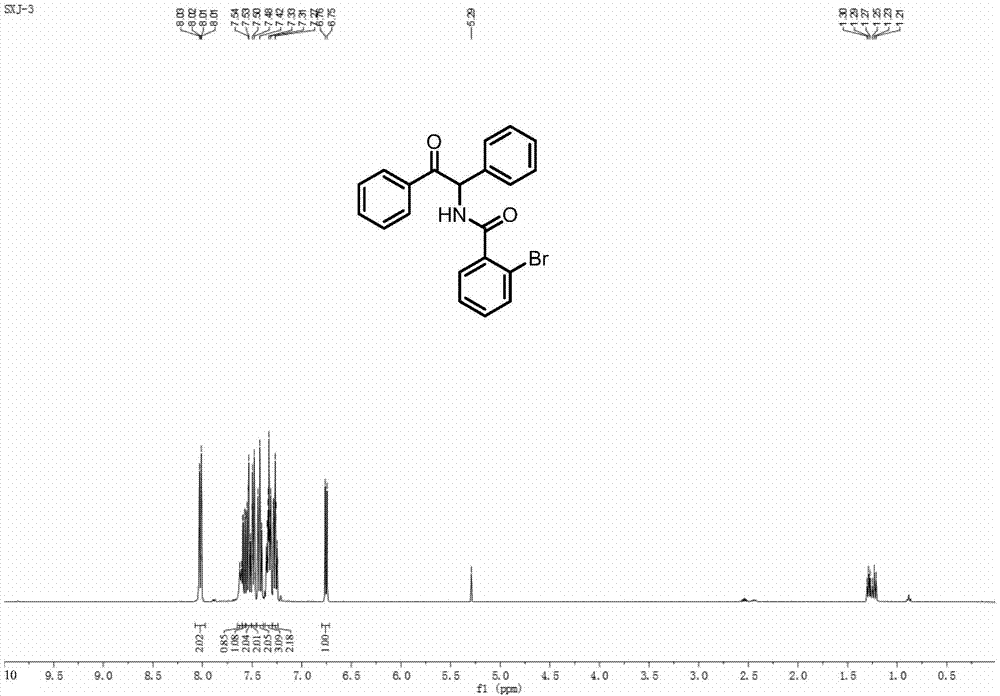
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率可达92%。
本实施例所获α-胺基酮衍生物3aa的核磁数据如下:
2-bromo-n-(2-oxo-1,2-diphenylethyl)benzamide(3aa).白色固体(45.3mg,产率92%).展开剂为石油醚/乙酸乙酯=4:1.1hnmr(400mhz,cdcl3)δ=8.02(dd,j=5.2,3.4,2h),7.62(d,j=7.0,1h),7.58(dd,j=7.9,1.1,1h),7.56–7.51(m,2h),7.51–7.46(m,2h),7.42(dd,j=10.6,4.8,2h),7.37–7.30(m,3h),7.30–7.24(m,2h),6.76(d,j=7.2,1h).13cnmr(101mhz,cdcl3)δ=195.2,166.5,137.0,136.8,134.2,133.9,133.5,131.5,129.9,129.2,128.8,128.5,128.4,127.5,124.4,119.6,59.2.hrmscalculatedforc21h17brno2+,394.0443,found394.0420.
实施例2
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(251.3mg,1.25mmol)、2h-氮杂环丙烷化合物2a(241.6mg,1.25mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率最高可达30%。
实施例3
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(20.1mg,0.1mmol)、2h-氮杂环丙烷化合物2a(24.6mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率最高可达85%。
实施例4
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(1.26g,6.25mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率最高可达19%。
实施例5
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(2.42g,1.25mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3aa,展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率42%。
实施例6
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(0.1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3aa,展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率58%。
实施例7
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(50.2mg,0.25mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3aa,展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率最高可达68%。
实施例8
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(66mg,1equiv)、dmap(1equiv)、li2co3(0.1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3aa,展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率50%。
实施例9
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(50.2mg,0.25mmol)、2h-氮杂环丙烷化合物2a(48.4mg,0.25mmol)、二氧六环(2ml),升温至80℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率仅有73%。
实施例10
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至80℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物(3aa),展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率仅有38%。
实施例11
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,0.0125mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应12小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3aa,展开剂为石油醚/乙酸乙酯=4:1,通过核磁和高分辨质谱方法确定其结构,参见图2、图3和图10所示,产率最高可达80%。
实施例12
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1b(15.3mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ba,通过核磁和高分辨质谱方法确定其结构,参见图4、图5和图11所示,产率最高可达70%。
本实施例所获α-胺基酮衍生物3ba的核磁数据如下:
n-(2-oxo-1,2-diphenylethyl)benzamide(3ba).白色固体(27.6mg,产率70%).展开剂为石油醚/乙酸乙酯=10:1.1hnmr(600mhz,cdcl3)δ8.10–7.99(m,2h),7.91–7.81(m,2h),7.74(d,j=6.7hz,1h),7.57–7.47(m,4h),7.43(m,3h),7.32(t,j=7.6hz,2h),7.28–7.23(m,1h),6.76(d,j=7.0hz,1h).13cnmr(151mhz,cdcl3)δ=195.8,166.3,137.3,134.2,133.9,133.9,131.8,129.3,129.2,128.8,128.6,128.4,128.3,127.2,58.9.hrmscalculatedforc21h18no2+,316.1338,found316.1297.
实施例13
本实施例的反应合成步骤及产物结构式如下:
本实施例的具体操作步骤如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类类化合物1c(20.9mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ca,通过核磁和高分辨质谱方法确定其结构,参见图6、图7和图12所示,产率最高可达92%。
本实施例所获α-胺基酮衍生物3ca的核磁数据如下:
3-nitro-n-(2-oxo-1,2-diphenylethyl)benzamide(3ca).黄色固体(41.4mg,产率92%).1hnmr(400mhz,cdcl3)δ=8.68(t,j=1.9,1h),8.36(m,1h),8.23–8.14(m,1h),8.06–7.99(m,2h),7.85(d,j=6.8,1h),7.65(t,j=8.0,1h),7.59–7.52(m,1h),7.52–7.47(m,2h),7.43(dd,j=10.6,4.8,2h),7.39–7.32(m,2h),7.32–7.24(m,1h),6.74(d,j=6.9,1h).13cnmr(101mhz,cdcl3)δ=195.3,164.0,148.3,136.7,135.7,134.1,134.0,133.1,129.8,129.4,129.2,128.9,128.8,128.4,126.3,122.3,59.3.hrmscalculatedforc21h17n2o4+,361.1188,found361.1189.
实施例14
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1d(22.0mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、dioxane(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3da,通过核磁和高分辨质谱方法确定其结构,参见图8、图9和图13所示,产率最高可达63%。
本实施例所获α-胺基酮衍生物3da的核磁数据如下:
2,3,4-trifluoro-n-(2-oxo-1,2-diphenylethyl)benzamide(3da).白色固体(29.1mg,产率63%).展开剂为石油醚/乙酸乙酯=4:1.1hnmr(400mhz,cdcl3)δ=8.31–8.17(m,1h),8.10–7.98(m,2h),7.85(m,1h),7.60–7.54(m,1h),7.51(dd,j=5.2,3.3,2h),7.45(dd,j=10.6,4.8,2h),7.39–7.33(m,2h),7.33–7.26(m,1h),7.09(m,1h),6.74(dd,j=6.7,1.7,1h).13cnmr(101mhz,cdcl3)δ=194.9,172.7,160.6(dd,j=4.5,1.7hz),148.6,144.7,136.7,134.0,134.0,129.3,129.2,128.8,128.6,128.4,125.8(dd,j=8.4,4.2hz),124.848,112.738(dd,j=17.4,3.5hz),59.48.19fnmr(376mhz,cdcl3)δ-127.93(dd,j=20.1,11.3hz),-134.19(dd,j=21.8,11.3hz),-159.45(t,j=21.0hz).hrmscalculatedforc21h15f3no2+,370.1055,found370.1070.
实施例15
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1e(16.0mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ea,通过核磁和高分辨质谱方法确定其结构,产率最高可达67%。
本实施例所获α-胺基酮衍生物3ea的核磁数据如下:
n-(2-oxo-1,2-diphenylethyl)thiophene-2-carboxamide(3ea).白色固体(26.9mg,产率67%).1hnmr(400mhz,cdcl3)δ=8.06–7.97(m,2h),7.63–7.51(m,3h),7.51–7.38(m,5h),7.36–7.29(m,2h),7.29–7.22(m,2h),7.08(dd,j=4.9,3.8,1h),6.71(d,j=7.0,1h).13cnmr(101mhz,cdcl3)δ=195.6,160.9,144.5,138.5,137.1,134.2,133.9,130.4,129.3,129.2,128.8,128.5,128.4,127.6,58.8.hrmscalculatedforc19h16no2s+,322.0902,found322.0882.
实施例16
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1f(7.5mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3fa,通过核磁和高分辨质谱方法确定其结构,产率最高可达60%。
本实施例所获α-胺基酮衍生物3fa的核磁数据如下:
n-(2-oxo-1,2-diphenylethyl)acetamide(3fa).白色固体(19.0mg,产率60%).1hnmr(400mhz,cdcl3)δ=8.01–7.93(m,2h),7.55–7.48(m,1h),7.44–7.36(m,4h),7.34–7.24(m,3h),6.97(d,j=6.7,1h),6.58(d,j=7.4,1h),2.05(s,3h).13cnmr(101mhz,cdcl3)δ=195.9,169.2,137.3,134.3,133.8,129.2,129.1,128.7,128.4,128.2,58.5,23.3.hrmscalculatedforc16h16no2+,254.1181,found254.1180.
实施例17
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1g(14.3mg,0.125mmol)、2h-氮杂环丙烷化合物2a(24.2mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ga,通过核磁和高分辨质谱方法确定其结构,产率最高可达93%。
本实施例所获α-胺基酮衍生物3ga的核磁数据如下:
2,2,2-trifluoro-n-(2-oxo-1,2-diphenylethyl)acetamide(3ga).白色固体(35.7mg,收率93%).1hnmr(400mhz,cdcl3)δ=8.04–7.88(m,3h),7.55(t,j=7.4,1h),7.46–7.38(m,4h),7.38–7.27(m,3h),6.49(d,j=7.0,1h).13cnmr(101mhz,cdcl3)δ=193.7,156.3(q,j=37.8hz),147.1,135.3,134.4,133.4,129.5,129.3,129.2,128.9,128.3,115.7(d,j=287.8hz),58.9.19fnmr(376mhz,cdcl3)δ=-75.73.hrmscalculatedforc16h13f3no2+,308.0898,found308.0896.
实施例18
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2b(26.4mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ab,通过核磁和高分辨质谱方法确定其结构,产率最高可达92%。
本实施例所获α-胺基酮衍生物3ab的核磁数据如下:
2-bromo-n-(1-(4-fluorophenyl)-2-oxo-2-phenylethyl)benzamide(3ab).白色固体(47.4mg,收率92%).1hnmr(400mhz,cdcl3)δ=8.06–8.00(m,2h),7.70(d,j=6.8,1h),7.64–7.54(m,3h),7.54–7.43(m,4h),7.38(td,j=7.5,1.2,1h),7.32(dd,j=7.7,1.8,1h),7.09–7.01(m,2h),6.79–6.73(m,1h).13cnmr(101mhz,cdcl3)δ=195.1,166.5,162.62(d,j=248.1hz),136.9,134.1,134.0,134.0,133.6,132.75(d,j=3.1hz),131.5,130.24(d,j=8.4hz),129.8,129.03(d,j=32.8hz),127.5,119.6,116.21(d,j=21.8hz),58.4.19fnmr(376mhz,cdcl3)δ=-112.85.hrmscalculatedforc21h16brfno2+,412.0348,found412.0356.
实施例19
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2c(28.5mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ac,通过核磁和高分辨质谱方法确定其结构,产率最高可达83%。
本实施例所获α-胺基酮衍生物3ac的核磁数据如下:
2-bromo-n-(1-(4-chlorophenyl)-2-oxo-2-phenylethyl)benzamide(3ac).白色固体(44.5mg,产率83%).1hnmr(400mhz,cdcl3)δ=8.02(d,j=7.9,2h),7.73(d,j=6.7,1h),7.64–7.52(m,3h),7.46(dd,j=7.6,4.2,4h),7.40–7.26(m,4h),6.74(d,j=6.9,1h).13cnmr(101mhz,cdcl3)δ=194.9,166.5,136.8,135.4,134.5,134.2,133.9,133.6,131.6,129.9,129.8,129.4,129.2,128.9,127.5,119.6,58.4.hrmscalculatedforc21h16brclno2+,430.0032,found430.0034.
实施例20
本实施例的反应合成步骤及产物结构式如下:
于反应容器内加入pd(oac)2(2.8mg,mmol)、氯[1,3-双(2,6-二异丙基苯基)咪唑-2-亚基]银(6.6mg,10mol%)、dmap(1equiv)、li2co3(1equiv)、羧酸类化合物1a(25.1mg,0.125mmol)、2h-氮杂环丙烷化合物2d(25.9mg,0.125mmol)、二氧六环(2ml),升温至110℃,反应18小时。将反应液冷却至室温,减压除去溶剂后,柱层析分离得到相应的α-胺基酮衍生物3ad,通过核磁和高分辨质谱方法确定其结构,产率最高可达85%。
本实施例所获α-胺基酮衍生物3ad的核磁数据如下:
2-bromo-n-(2-oxo-1-phenyl-2-(p-tolyl)ethyl)benzamide(3ad).白色固体(43.4mg,产率85%).1hnmr(400mhz,cdcl3)δ=8.02(d,j=7.5,2h),7.64–7.49(m,4h),7.37(ddd,j=16.6,11.4,7.3,5h),7.29–7.23(1h),7.13(d,j=7.9,2h),6.72(d,j=7.2,1h),2.28(s,3h).13cnmr(101mhz,cdcl3)δ=195.3,166.4,138.4,137.1,134.3,133.8,133.8,133.5,131.4,129.9,129.9,129.2,128.8,128.3,127.5,119.6,58.9,21.2.
对照例1
本对照例与实施例1基本一致,不同之处在于:反应温度为40℃,几乎未得到相应产物。
对照例2
本对照例与实施例1基本一致,不同之处在于:反应温度为室温,几乎未得到相应产物。
此外,本案发明人还参照实施例1-实施例20的方式,以本说明书中列出的其它原料和条件等进行了试验,并同样成功合成了α-胺基酮衍生物。
综上所述,本发明首次使用2h-氮杂环丙烷类化合物作为合成子,经由羧酸对2h-氮杂环丙烷化合物的亚胺双键进行亲核加成反应、开环重排反应,成功一步构建α-胺基酮衍生物;本发明对水和空气稳定不敏感,操作简单,反应条件温和,反应效率高,底物的适用范围广,原子利用率高,反应区域选择性和化学选择性高,所制备的α-胺基酮衍生物结构新颖,具有潜在的应用价值。
应当理解,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!