一种可溶性氨基化刚性棒状聚合物及其制备方法和组合物与流程
本发明涉及高分子聚合物领域,具体涉及一种可溶的氨基化刚性棒状聚合物、其组合物及其纤维增强复合材料。
背景技术:
分子复合材料是指以刚性棒状大分子作为增强相,柔性聚合物为基体的复合材料。与传统纤维增强高分子复合材料使用微米级纤维(如玻璃纤维、碳纤维、聚合物纤维等)实现增强不同,分子复合材料通过将刚性棒状大分子以分子水平均匀分散在柔性聚合物基体中实现增强。由于刚性棒状大分子具有伸直链的分子结构并具有力学性能优异(高强、高模)、耐热性好等优点,从理论上讲,如果能使其以分子水平分散在基体中将具有很大的长径比和极大的比表面积,从而成为理想的增强体,同时还具有低粘度和良好的加工性能。分子复合材料甚至被认为有可能获得比短纤维增强复合材料更高的模量和强度。
takayanagi等采用共沉淀法制备了尼龙66/聚对苯二甲酰对苯二胺(pa66/ppta)以及pa6/ppta分子复合材料,复合材料的断面可以看到ppta微纤拔出,其尺寸大约为10-30nm,并且均匀地分散在基体中。同时,ppta的加入使力学性能大幅度提高。当添加量为5wt%时,增强效果相当于40wt%填充量的玻璃纤维纤维增强尼龙。此外,测试结果表明ppta可以使得诱导基体结晶,使熔点及结晶度升高,对耐热性的提高也有帮助(uetas,sakamotot,takayanagim.effectsofmolecular-weightofnylon-6onthestructureandpropertiesofnylon-6/poly(p-phenyleneterephthalamide)molecularcomposites.polymerjournal[j]1993,25,31-40)。
ueta等为制备ppta及环氧树脂分子复合材料,用环氧树脂取代ppta大分子酰胺键上的氢原子,得到环氧树脂接枝的ppta,与环氧树脂混合后制备出分子复合材料,发现弯曲模量及弯曲强度大幅度提高,分别为环氧树脂的1.66及1.36倍(uetas,leiwy,kogak,takayanagim.preparationofn-graftedpoly(p-phenyleneterephthalamide)andapplicationstoamolecularcompositewithepoxy-resin.polymerjournal[j]1993,25,185-191)。
因此ppta及环氧树脂接枝共聚物的引入使得分子复合材料相容性大幅度提高,力学性能增强。但该技术存在以下缺点:首先,该接枝反应使用氢化钠为催化剂,危险性大;第二,更重要的是酰胺键上的氢原子被环氧树脂取代后,ppta大分子链发生了扭曲,棒状结构被破坏,因此增强幅度无法继续提高。
专利us20060058469、us20030166796、us20030166796、us20160194542a1、us20100096169、us20090081466和cn200780045284报道了酚羟基取代聚酰胺及其与橡胶的共聚物用于环氧树脂改性,us20160194542a1和us20130105200报道了酚羟基取代聚酰亚胺,其热固性树脂具有较高的耐热性、柔韧性、粘接性、电绝缘性以及阻燃性。
上述酚羟基取代聚酰胺和聚酰亚胺均大分子量采用无规线团结构,而非伸直链型的取代聚酰胺和聚酰亚胺,无规线团型大分子比较易于溶解于树脂中,因此将酚羟基取代聚合物作为添加剂直接与环氧树脂混合使用。
但是,上述方法不适合伸直链型的取代聚酰胺和聚酰亚胺,因为伸直链型聚酰胺和聚酰亚胺之间的氢键作用强,即使有酚羟基等取代基团,也难以直接在树脂中均匀分散和溶解。同时,由于酚羟基与环氧树脂在室温下能够发生反应,直接将酚羟基取代伸直链型聚酰胺、聚酰亚胺与环氧树脂混合会发生自交联反应,因此在分散过程中易产生凝胶,均匀性不理想,并且稳定存储时间短,此外,上述专利仅仅提高了树脂的粘接和耐热等性能。
技术实现要素:
本发明的目的在于提供了一种具有极高反应活性的、溶解性能好的氨基化刚性棒状聚合物及其制备方法,还提供了一种由所述氨基化刚性棒状聚合物与热固性树脂组成的组合物,大大提高了热固性树脂的强度和韧性。
本发明提供了一种可溶性氨基化刚性棒状聚合物,包括结构通式i的氨基化聚酰胺:
或结构通式ii的氨基化聚酰亚胺:
或结构通式iii的氨基化聚酰胺酰亚胺:
其中,ar1、ar2、ar3、ar4、ar5、ar7选自苯基、联苯基、萘基、吡啶基、联吡啶基,其中,至少一种选自苯基、联苯基或萘基;ar8选自苯基、联苯基、萘基、二苯甲酮或二苯醚基团;
以上通式中,r1、r2、r3、r4、r5、r6选自氨基或甲基,其中,至少一种选自氨基;
以上通式中,i、j、i’、j’、i”、j”表示1到4的整数;
以上通式中,m、n、p和q为各重复结构单元的数量,为1~1000的整数,且m+n>1,p+q>1;
以上通式中,x、y为1~1000的整数;
通式(i)和(iii)中,(i+j+i’+j’)/(m+n)为0.1~2;
通式(ii)和(iii)中,(i+j)/(p+q)为0.1~1。
本发明还提供了一种上述氨基化聚酰胺、氨基化聚酰亚胺和氨基化聚酰胺酰亚胺的制备方法,所述的氨基化聚酰胺、氨基化聚酰亚胺或氨基化聚酰胺酰亚胺由硝基取代聚酰胺、硝基取代聚酰亚胺或硝基取代聚酰胺酰亚胺溶于有机溶剂后加入金属催化剂进行催化加氢还原反应得到。
所述的催化加氢还原反应的硝基转化率大于60%。
上述硝基转化率指催化加氢还原反应过程中,所述硝基取代聚酰胺、硝基取代聚酰亚胺和硝基取代聚酰胺酰亚胺中转化成氨基的硝基基团在全部硝基基团中所占的百分比;转化率低于60%不能保证聚合物在有机溶剂和热固性树脂中具有良好的溶解性和反应活性。
所述的金属催化剂选自钯碳催化剂、雷尼镍催化剂或氯化锡催化剂,优选为钯碳催化剂。
所述的有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或六甲基磷酰胺中的任意一种。
所述的催化加氢还原反应的反应温度为40℃-160℃且不超过所选有机溶剂的沸点,反应压力为0.1-5mpa,反应时间为0.5-24h。
所述的催化加氢还原反应的氢气源为氢气或水合肼。
所述的硝基取代聚酰胺、硝基取代聚酰亚胺和硝基取代聚酰胺酰亚胺中的硝基侧基与主链中苯环的摩尔比为1:1~5,当摩尔比小于1:5时,会使得到的产物不能在有机溶剂或树脂单体中充分溶解。
所述的硝基取代聚酰胺由芳香族二酸或芳香族二酰氯与芳香族二胺通过溶液缩聚得到;
其中,所述芳香族二酸选自通式(1)-(4)中的一种或多种,优选为对苯二甲酸、硝基对苯二甲酸、2,5-二硝基对苯二甲酸、萘二甲酸、硝基-2,6-萘二甲酸、二硝基-2,6-萘二甲酸、联苯二甲酸、2-硝基联苯二甲酸、2,2’-二硝基联苯二甲酸;
所述芳香族二酰氯选自所述芳香族二酸的酰氯化衍生物中的一种或多种;
所述的芳香族二胺选自通式(5)-(7)中的一种或多种,优选为对苯二胺、硝基对苯二胺、二硝基对苯二胺、萘二胺、单硝基萘二胺、二硝基萘二胺、二氨基联苯、二硝基二氨基联苯;
通式(1)-(7)中,r1选自硝基或甲基中的一种或两种;其中,至少一种选自硝基;
以上通式中,i和j为0~4的整数;
所述的芳香族二酸或二酰氯与芳香族二胺的摩尔比为1:(0.5~2);
所述的溶液缩聚反应为高温溶液缩聚反应、低温溶液缩聚反应或界面缩聚反应;
所述高温溶液缩聚反应的具体步骤为:在反应釜中加入有机溶剂、芳香族二酸、芳香族二胺、缩合剂、酸吸收剂和盐,加热至40~150℃反应2~48h,水洗、过滤、干燥,得到聚酰胺。
所述的芳香族二酸和芳香族二胺的摩尔比为(2:1)~(1:2),所述的芳香族二酸在有机溶剂中的质量浓度为0.1%~30%;
所述的有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或者六甲基磷酰胺;
所述的缩合剂为亚磷酸三苯酯或三硝基氯苯中的至少一种;
所述的酸吸收剂为吡啶、三乙基胺、2,4-二甲基吡啶或咪唑中的一种或多种,所述的酸吸收剂在溶液中的浓度为1~40%;
所述的盐为氯化锂、氯化钙或硫氰化钾,所述的盐在溶液中的浓度为0.1~20%。
所述低温溶液缩聚反应的具体步骤为:先将芳香族二酸用氯化亚砜或草酰氯进行酰氯化,得到芳香二酰氯;再将得到的芳香二酰氯、芳香族二胺、有机溶剂、酸吸收剂和盐加入反应釜中,在0~40℃搅拌反应2~48小时,水洗、过滤、干燥,得到聚酰胺。
所述的芳香族二酰氯和芳香族二胺的摩尔比为(2:1)~(1:2),所述的芳香族二酰氯在有机溶剂中的质量浓度为0.1%~30%;
所述的有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或六甲基磷酰胺;
所述酸吸收剂为吡啶、三乙基胺、2,4-二甲基吡啶或咪唑中的一种或多种,所述的酸吸收剂在溶液中的浓度为1~40%;
所述盐为氯化锂、氯化钙或硫氰化钾,所述盐在溶液中的浓度为0.1~20%。
所述界面缩聚反应的具体步骤为:将芳香族二酸用氯化亚砜或草酰氯进行酰氯化,得到芳香族二酰氯,再将芳香族二酰氯溶解在氯仿、苯、正己烷、四氯化碳或甲苯中,得到芳香族二酰氯溶液;将芳香族二胺溶解在氢氧化钠或氢氧化钾水溶液中,得到芳香族二胺溶液;
将所述芳香族二酰氯溶液与芳香族二胺溶液在0~40℃混合搅拌2~48h,再经过水洗、过滤、干燥,得到聚酰胺。
所述的芳香族二酰氯和芳香族二胺的摩尔比为(2:1)~(1:2),所述芳香族二酰氯在有机溶剂中的质量浓度为0.1%~30%,所述芳香族二胺在水中的质量浓度为0.1%~30%。
所述的硝基取代聚酰亚胺由芳香族二酸酐与芳香族二胺缩聚而成;
所述芳香族二酸酐为均苯四甲酸酐、1,4,5,8-萘四甲酸酐、4,4'-氧双邻苯二甲酸酐、4,4'-羰基二邻苯二甲酸酐、3,3',4,4'-二苯甲酮四酸二酐、联苯四酸二酐、苝四甲酸酐等芳香族四酸酐中的一种或者多种;
所述芳香族二胺选自硝基对苯二胺、二硝基对苯二胺、单硝基萘二胺、二硝基萘二胺、二硝基二氨基联苯;
所述芳香族二酐为通式(8)-(13)中任意一种或多种;所述芳香族二胺为通式(5)-(7)中任意一种或多种;
所述的芳香族二酐和芳香族二胺的摩尔比为1:(0.5~2)。
所述的硝基取代聚酰亚胺具体制备步骤为:
(1)在反应釜中加入有机溶剂、芳香族二酸酐和芳香族二胺,在5~50℃反应2~48h,得到聚酰胺酸;
(2)加入脱水剂醋酸酐和叔胺类催化剂,加热至100~180℃进行闭环反应,再经过水洗、过滤、干燥,得到聚酰亚胺。
步骤(1)中,所述芳香族二酐和芳香族二胺的摩尔比为(2:1)~(1:2);
步骤(2)中,所述叔胺类催化剂为吡啶、三乙胺;所述有机溶剂为二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或六甲基磷酰胺。
所述的硝基取代聚酰胺酰亚胺的制备方法为:将芳香族二酐与过量的芳香族二胺反应得到端氨基聚酰亚胺;再将得到的端氨基聚酰亚胺与芳香族二酸或芳香族二酰氯进行缩聚反应得到所述硝基取代聚酰胺酰亚胺;
所述芳香族二酐为通式(8)-(13)中任意一种或多种;所述芳香族二胺为通式(5)-(7)中任意一种或多种;
所述芳香族二酸选自通式(1)-(4)中的一种或多种;所述芳香族二酰氯选自所述芳香族二酸的酰氯化衍生物中的一种或多种;
所述芳香族二酐、芳香族二胺和芳香族二酸或芳香族二酰氯的比例为1:(1.01~2):(0.01~2)。
所述的硝基取代聚酰胺酰亚胺具体制备步骤为:
(1)制备氨基封端的聚酰胺链段;
所述氨基封端的聚酰胺链段的具体制备方法为:在反应釜中加入有机溶剂、芳香族二酸、芳香族二胺、缩合剂、酸吸收剂和盐,加热至40~150℃反应2~48h,得到氨基封端的聚酰胺溶液;其中,所述的芳香族二胺和芳香族二酸的摩尔比为(1.01:1)~(2:1),所述芳香族二酸在有机溶剂中的质量浓度为0.1%~30%;
或者,所述氨基封端的聚酰胺链段的具体制备方法为:先将芳香族二酸用氯化亚砜或草酰氯进行酰氯化,得到芳香二酰氯;再将得到的芳香二酰氯、芳香族二胺、有机溶剂、酸吸收剂和盐加入反应釜中,搅拌反应2~48小时,得到氨基封端的聚酰胺溶液;其中,所述芳香族二胺和芳香族二酰氯的摩尔比为(1.01:1)~(2:1),所述芳香族二酰氯在有机溶剂中的质量浓度为0.1%~30%;
(2)在步骤(1)得到的氨基封端的聚酰胺溶液中加入芳香族二酐和芳香族二胺,在5~50℃反应2~48h,得到聚酰胺酰胺酸;再加入脱水剂酸酐和叔胺类催化剂,加热至100~180℃进行闭环反应,再经过水洗、过滤、干燥,得到聚酰胺酰亚胺。
步骤(1)中,所述的有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或六甲基磷酰胺;
所述的芳香族二酸选自对苯二甲酸、硝基对苯二甲酸、2,5-二硝基对苯二甲酸、萘二甲酸、硝基-2,6-萘二甲酸、二硝基-2,6-萘二甲酸、联苯二甲酸、2-硝基联苯二甲酸、2,2’-二硝基联苯二甲酸;
所述芳香族二酰氯为上述芳香族二酸的酰氯化衍生物;
所述的芳香族二胺选自对苯二胺、硝基对苯二胺、二硝基对苯二胺、萘二胺、单硝基萘二胺、二硝基萘二胺、二氨基联苯、二硝基二氨基联苯;
所述缩合剂选自亚磷酸三苯酯或三硝基氯苯中的一种或多种;
所述酸吸收剂为吡啶、三乙基胺、2,4-二甲基吡啶或咪唑中的一种或多种,所述的酸吸收剂在溶液中的浓度为1~40%;
所述盐为氯化锂、氯化钙或硫氰化钾,所述盐在溶液中的浓度为0.1~20%。
步骤(2)中,所述芳香族二胺和芳香族二酐的摩尔比为(0:2)~(1:0.95);
所述叔胺类催化剂为吡啶、三乙胺;
所述芳香族二酐选自均苯四甲酸酐、1,4,5,8-萘四甲酸酐、4,4'-氧双邻苯二甲酸酐、4,4'-羰基二邻苯二甲酸酐、3,3',4,4'-二苯甲酮四酸二酐、联苯四酸二酐、苝四甲酸酐等芳香族四酸酐中的一种或多种;
所述芳香族二胺选自硝基对苯二胺、二硝基对苯二胺、单硝基萘二胺、二硝基萘二胺、二硝基二氨基联苯中的一种或者多种。
本发明还提供了一种可溶性氨基化刚性棒状聚合物的组合物,所述组合物包括氨基化刚性棒状聚合物和热固性树脂,所述氨基化刚性棒状聚合物和热固性树脂的质量比为0.1~90:100。
所述热固性树脂为环氧树脂、酚醛树脂、苯并噁嗪树脂、乙烯基树脂、不饱和聚酯、热固性聚酰亚胺。
所述组合物由氨基化刚性棒状聚合物与热固性树脂进在有机溶剂中混合后,再经过固化得到;
所述有机溶剂为低沸点有机溶剂或水溶性高沸点有机溶剂;
所述低沸点有机溶剂为乙醇、甲醇、丙酮或四氢呋喃;所述水溶性高沸点有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氧六环、环丁砜、n-甲基吡咯烷酮或六甲基磷酰胺。
所述环氧树脂选自双酚a缩水甘油醚型环氧树脂、双酚f缩水甘油醚型环氧树脂、酚醛环氧树脂、缩水甘油酯型环氧树脂和缩水甘油胺型环氧树脂中的一种或多种。
所述双酚a缩水甘油醚型环氧树脂选自e-55、e-51、e-44、e-42或e-35;
所述双酚f缩水甘油醚型环氧树脂选自cydf-170、cydf-180(岳阳石化)、cydf-175(巴陵石化)或npef-170(台湾南亚);
所述酚醛环氧树脂选自f-51、f-44或f-42;
所述缩水甘油酯型环氧树脂选自tde-85;
所述缩水甘油胺型环氧树脂选自n,n,n’,n’-四缩水甘油基-4,4’-二胺基二苯甲烷(ag-80)、n,n,n’,n’-四缩水甘油基-4,4’-二胺基二苯醚或n,n,n’,n’-四缩水甘油基间二甲苯二胺。
氨基化刚性棒状聚合物中的氨基具有良好的反应活性,其可以与环氧基团进行接枝反应,也可以与双键进行迈克尔加成反应,从而与所述热固性树脂形成共价键连接。
本发明与现有技术相比,具有以下有益效果:
(1)本发明得到的氨基化刚性棒状聚合物主链具有伸直链(棒状)结构,聚合物中所含氨基使其具有较高的反应活性,其溶解性能、强度和刚性也很好;
(2)本发明利用氨基的高反应活性,将得到的氨基化刚性棒状聚合物与热固性树脂进行混合、反应,可以大大改善热固性树脂的强度和韧性,用途广泛;
(3)本发明得到的氨基化刚性棒状聚合物可以快速溶解于热固性树脂中,分散过程中不产生凝胶,加热固化过程中其参与环氧树脂的固化反应嵌入热固性树脂的三维交联网络中,使得到的组合物具有优异的存储稳定性;
(4)本发明得到的氨基化刚性棒状聚合物在极低的添加量下就能够同时实现环氧树脂的增强、增韧,克服了传统纳米增强技术增强程度低,并且同时使韧性降低的缺点;当聚合物的质量浓度为1wt%就可以使得到的环氧树脂固化物的拉伸强度>100mpa、弯曲强度>170mpa、断裂伸长率提高15%以上,明显高于纯环氧树脂固化物,也高于现有技术中所述的分子复合材料以及氧化石墨、碳纳米管或纳米粘土改性的环氧树脂固化物;
(5)本发明方法合成条件温和,不需要使用氢化钠一类易燃易爆的催化剂。
附图说明
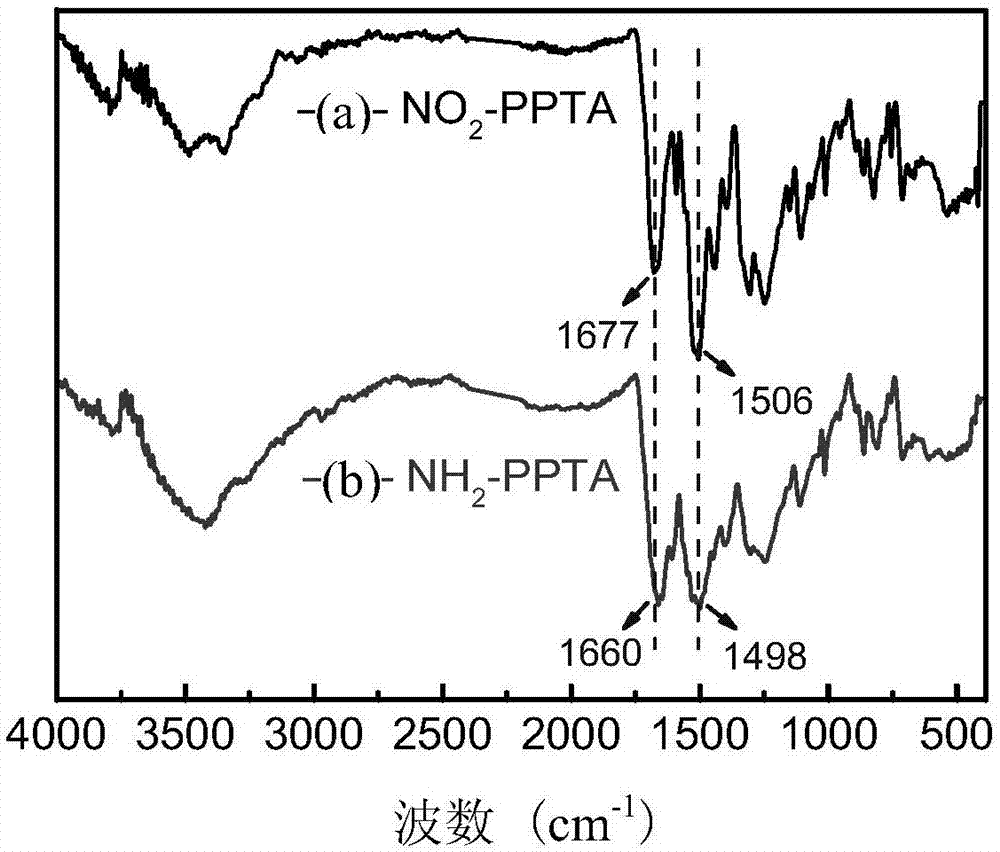
图1为实施例1制备的硝基取代聚酰胺(no2-ppta)和氨基取代聚酰胺(nh2-ppta)的红外谱图;
图2为实施例1制备的氨基取代聚对苯二甲酸对苯二胺(nh2-ppta)的环氧树脂的拉伸应力-应变曲线。
具体实施方式
下面结合实施例和比较例对本发明进行进一步说明,但本发明不限于这些实施例。固化后材料用万能电子试验机按照标准astmd-638测试拉伸性能,按照标准astmd-790弯曲性能。
实施例1-5
低温溶液缩聚反应:
(1)分别按下表1中指定的单体及其含量将溶剂、酸吸收剂、盐和二胺加入反应釜中并搅拌均匀,再加入二酰氯在0~40℃反应搅拌反应一段时间,加水沉淀、水洗、过滤、干燥,得到硝基取代聚酰胺;
(2)将10g步骤(1)得到的硝基取代聚酰胺重新溶解在500ml的dmac中,再加入0.1g钯碳催化剂,在160℃、3mpa下催化加氢0.5h,冷却、加水沉淀、过滤、干燥,得到氨基取代聚酰胺。
实施例1中步骤(1)得到的硝基取代聚酰胺的红外谱图如图1(a)所示,1677cm-1处为酰胺键的吸收峰,1506cm-1处为硝基的特征峰,证明了硝基取代聚酰胺的化学结构;实施例1中步骤(2)得到的氨基取代聚酰胺的红外谱图如图1(b)所示,硝基的特征峰(1506cm-1)明显减小,证明硝基被还原成了氨基。
将实施例1得到的氨基取代聚酰胺(nh2-ppta)分别按照质量浓度0.1%、0.3%、0.5%、0.7%、0.9%、1.1%、1.3%与400gafg-90和200g固化剂二甲硫基甲苯二胺混合搅拌均匀,然后浇注到模具中分别在120℃、160℃和220℃加热2.5h进行固化,得到了氨基取代聚酰胺与afg-90环氧树脂复合得到的复合材料。
图2为实施例1中得到的复合材料的拉伸应力应变曲线,由图2可知,氨基取代聚酰胺的添加量仅为0.5-0.9%时,复合材料的拉伸强度就达到了100.2mpa,相对应的断裂伸长率为7%,分别比纯树脂提高了82%和32%。
实施例2-5得到的复合材料性能均高于纯树脂,最大拉伸强度大于100mpa,断裂伸长率比纯树脂提高了15%以上,弯曲强度大于170mpa。
实施例6-15
高温溶液缩聚反应:
(1)分别按下表1中指定的单体及其含量将溶剂、酸吸收剂、缩合剂、盐和二胺加入反应釜中并搅拌均匀,再加入芳香族二酸在60~160℃搅拌、反应一段时间,加水沉淀、水洗、过滤、干燥,得到硝基取代聚酰胺;
(2)将10g步骤(1)得到的硝基取代聚酰胺溶解在500ml的dmac中,再加入0.1g钯碳催化剂,在100℃、0.1mpa下催化加氢24h,冷却、加水沉淀、过滤、干燥,得到氨基取代聚酰胺。
将实施例6-15得到的氨基取代聚酰胺各取2g分别与100g双酚a型环氧树脂e-44混合搅拌后,再加入50g固化剂二氨基二苯砜搅拌均匀,然后浇注到模具中,在140℃加热16h进行固化,得到复合材料。
测得实施例6-15得到的复合材料的拉伸强度大于100mpa、断裂伸长率比纯树脂提高15%以上、弯曲强度大于170mpa。
实施例16-18
界面缩聚反应:
(1)分别按下表1中指定的单体及其含量将水和二胺的盐酸盐加入反应釜中搅拌均匀,再将二酰氯溶解于有机溶剂中加入反应釜,在0~40℃搅拌反应一段时间,加水沉淀、水洗、过滤、干燥,得到硝基取代聚酰胺;
(2)将1~40g步骤(1)得到的硝基取代聚酰胺溶解在100ml的n-甲基吡咯烷酮中,再加入0.5g雷尼镍催化剂,在100℃、氢气为氢源、1mpa条件下催化加氢24h,冷却、加水沉淀、过滤、干燥,得到氨基取代聚酰胺。
将实施例16-18得到的氨基取代聚酰胺按照0.1%~10%比例加入400g缩水甘油胺型环氧树脂afg-90,充分搅拌后,水洗三遍、80℃真空干燥12h后,加入400g固化剂e100,搅拌均匀,然后浇注到模具中在140℃加热16h进行固化,得到复合材料;
测得实施例16-18得到的复合材料的拉伸强度均在100~120mpa之间、断裂伸长率比纯环氧树脂提高了20%以上、弯曲强度大于170mpa。
表1
注:dmf:n,n-二甲基甲酰胺;dmac:n,n-二甲基乙酰胺;dmso:二甲基亚砜;doa:二氧六环;tms:环丁砜;nmp:n-甲基吡咯烷酮;hmpa:六甲基磷酰胺;py:吡啶;teae:三乙胺。tpc:对苯二甲酰氯;tpa:对苯二甲酸;ndca:2,6-萘二甲酸;bpdca:2,2'-联吡啶-4,4'-二甲酸;ntpa:2-硝基对苯二甲酸;n-2,6-ndca:硝基-2,6-萘二甲酸;dn-2,6-ndca:硝基-2,6-萘二甲酸;2,5-dntpa:2,5-二硝基对苯二甲酸;bpda:4,4'-联苯二甲酸;2-nbpda:2-硝基联苯二甲酸;dnbpda:2,2’-二硝基联苯二甲酸;ppd:对苯二胺;ndpa:2-硝基对苯二胺;nda:1,5-萘二胺;nnda:单硝基萘二胺;dnnda:二硝基萘二胺;bzd:二氨基联苯;dnbzd:二硝基二氨基联苯;tpp:亚磷酸三苯酯;pc:三硝基氯苯。
实施例19-25
(1)分别按下表2中指定的单体及其含量将溶剂和芳香二胺搅拌均匀,将芳香二酸酐单体加入反应釜搅拌反应,得到聚酰胺酸;再加入脱水剂醋酸酐和催化剂,加热至100-180℃进行闭环反应,再经过水洗、过滤、干燥,得到硝基取代聚酰亚胺;
(2)取10g步骤(1)得到的硝基取代聚酰亚胺溶解在500ml的dmac中,再加入钯碳催化剂,在100℃、3mpa的条件下催化加氢反应24h,冷却、加水沉淀、过滤、干燥,得到氨基化聚酰亚胺。
将实施例19-25得到的氨基化聚酰亚胺各取10g溶解在100ml的n-甲基吡咯烷酮中,加入100g甲基丙烯酸缩水甘油酯,充分搅拌后,水洗三遍、乙醇洗三遍,60℃真空干燥12h后,得到侧基带有甲基丙烯酸酯基团的可溶聚酰亚胺大分子;
将1-50g上述刚性聚合物加入400g乙烯基树脂(商品牌号411),适量固化剂、促进剂,搅拌均匀,然后浇注到模具中在140℃加热2h进行固化,得到复合材料。
测得实施例19-25得到的复合材料的拉伸强度均大于100mpa,断裂伸长率大于6%,比纯树脂提高20%以上,弯曲强度大于170mpa。
表2
实施例26
(1)以0.05moltpc和0.1molndpa按实施例1所述步骤合成氨基封端硝基取代聚酰胺,再加入0.05mol均苯四甲酸酐按照实施例19所述步骤完成聚酰亚胺链段的合成;
(2)取10g得到的硝基取代聚酰胺聚酰亚胺溶解在500ml的dmac中,用钯碳催化剂在100℃、3mpa下催化加氢24h,冷却、加水沉淀、过滤、干燥,得到氨基化聚酰胺聚酰亚胺;
经过核磁共振氢谱测试,硝基转化率约为80%。
取10g得到的氨基化聚酰胺聚酰亚胺溶解在100ml的n-甲基吡咯烷酮中,加入50g双酚a型环氧树脂e-51,充分搅拌后,水洗三遍、80℃真空干燥12h后,取1g聚合物加入100g苯并噁嗪树脂(牌号:aibz321)的乙醇溶液(浓度30%),搅拌均匀,然后浇注到模具中除去乙醇后在160℃加热4h进行固化,得到复合材料。
测得得到的复合材料的拉伸强度大于100mpa,断裂伸长率大于6%,比纯树脂提高20%以上,测得弯曲强度大于170mpa。
实施例27
合成聚酰胺酰亚胺:首先以0.05moltpc和0.05molndpa和0.05moldnnda按实施例2的条件合成氨基封端聚酰胺,然后加入0.05mol4,4'-氧双邻苯二甲酸酐按照实施例20的条件完成聚酰亚胺链段的合成。
按照实施例26的条件进行催化加氢,加入环氧树脂并固化。
测得得到的复合材料的拉伸强度大于100mpa,断裂伸长率大于6%,比纯树脂提高20%以上,测得弯曲强度大于170mpa。
实施例28
合成聚酰胺酰亚胺:首先以0.09moltpc和0.1moldnbzd按实施例5合成氨基封端聚酰胺,然后加入0.01mol联苯四酸二酐按照实施例21的条件完成聚酰亚胺链段的合成。
按照实施例26的条件进行催化加氢,加入环氧树脂并固化。
测得得到的复合材料的拉伸强度大于100mpa,断裂伸长率大于6%,比纯树脂提高20%以上,测得弯曲强度大于170mpa。
对比例1
60g双酚a型环氧树脂e-51,加入40g固化剂四氢邻苯二甲酸酐,搅拌均匀,然后浇注到模具中在140℃加热16h进行固化;
测得拉伸强度均为64mpa,断裂伸长率5%,弯曲强度为110mpa。
对比例2
60g缩水甘油胺环氧树脂afg-90,加入30g固化剂二乙基甲苯二胺,搅拌均匀,然后浇注到模具中在140℃加热2h,180℃加热2h最后在220℃加热2h完成固化;
测得拉伸强度均为55mpa,断裂伸长率4%。弯曲强度为124mpa。
对比例3
50g缩水甘油胺环氧树脂ag-80,加入20g固化剂二氨基二苯砜,溶解并搅拌均匀,然后浇注到模具中在130℃加热2h,160℃加热2h最后在220℃加热2h完成固化;
测得拉伸强度均为80mpa,断裂伸长率4%。弯曲强度为130mpa。
对比例4
50g缩水甘油胺环氧树脂ag-80,加入20g固化剂二氨基二苯砜,溶解并搅拌均匀,然后浇注到模具中在130℃加热2h,160℃加热2h最后在220℃加热2h完成固化;
测得拉伸强度均为80mpa,断裂伸长率4%,弯曲强度为130mpa。
通过上述实施例和比较例可以看出本发明的氨基化可溶钢性棒状聚合物能够有效地提高热固性树脂的力学性能。
- 还没有人留言评论。精彩留言会获得点赞!