一种检测半胱氨酸的新型荧光探针及其制备方法和应用与流程
本发明属于分析化学技术领域,尤其涉及一种检测半胱氨酸的新型荧光探针及其制备方法和应用。
背景技术:
生物硫醇如半胱氨酸(cys),谷胱甘肽(gsh),同型半胱氨酸(hcy)在生理和病理过程中起至关重要的作用。然而,细胞内硫醇水平的改变与很多疾病密切相关。体内缺乏半胱氨酸会导致多种病症,如儿童生长缓慢,肝损伤和皮肤损伤等。谷胱甘肽(gsh)在细胞内含量在1mm到15mm之间,是细胞内最富裕的硫醇,在维持细胞的氧化还原动态平衡中起着重要作用。血液中同型半胱氨酸的浓度增加会导致维生素b12的缺失和老年痴呆症。由于半胱氨酸,谷胱甘肽和同型半胱氨酸具有不同生理和病理功能,因此,检测半胱氨酸而不受谷胱甘肽和同型半胱氨酸显得尤为重要。
开发检测半胱氨酸的荧光探针越来越受到重视,由于其具有选择性好、灵敏度高、快速简便等优点。
技术实现要素:
本发明要解决的技术问题是克服现有技术的不足,提供一种选择性好、灵敏度高、快速简便的检测半胱氨酸的新型荧光探针,还相应提供该检测半胱氨酸的新型荧光探针的制备方法及应用。
为解决上述技术问题,本发明采用以下技术方案:
一种检测半胱氨酸的新型荧光探针,所述检测半胱氨酸的新型荧光探针为4-(7-二乙氨基香豆素-3-乙烯基)-1-(4-丙烯酰氧基)苄基吡啶溴盐,结构式如式(1):
作为一个总的发明构思,本发明还提供一种上述的检测半胱氨酸的新型荧光探针的制备方法,包括以下步骤:
(1)对羟基苄醇与丙烯酰氯反应,得到4-(羟甲基)丙烯酸苯酯,结构式如式(4):
(2)步骤(1)所得的4-(羟甲基)丙烯酸苯酯与三溴化磷反应,得到4-(溴甲基)丙烯酸苯酯,结构式如式(3):
(3)4-二乙氨基水杨醛与丙二酸二乙酯反应,得到7-二乙氨基香豆素,结构式如式(6):
(4)步骤(3)所得的7-二乙氨基香豆素进行维尔斯迈尔(vilsmeier)反应,得到7-二乙氨基香豆素-3-甲醛,结构式如式(5):
(5)步骤(4)所得的7-二乙氨基香豆素-3-甲醛与4-甲基吡啶通过脱水缩合反应,得到7-二乙氨基-3-(4-吡啶基)乙烯基香豆素,结构式如式(2):
(6)步骤(2)所得的4-(溴甲基)丙烯酸苯酯与步骤(5)所得的7-二乙氨基-3-(4-吡啶基)乙烯基香豆素反应,得到式(1)所示的化合物。
优选的,所述步骤(1)的具体过程为:
将碳酸钾溶于丙酮和水的混合溶液中(丙酮和水的体积比4:1),再加入丙烯酰氯,冷却至0℃,控温0℃缓慢滴加对羟基苄醇溶液,滴加完后控温0℃反应4~6h,反应完后,用饱和氯化钠溶液淬灭,用二氯甲烷萃取,有机相用无水硫酸钠干燥,蒸发溶剂后,过硅胶色谱柱纯化,得产物为4-(羟甲基)丙烯酸苯酯。
优选的,所述对羟基苄醇与丙烯酰氯的摩尔比为1∶0.8~1.0。
优选的,所述步骤(2)的具体过程为:
将4-(羟甲基)丙烯酸苯酯用二氯甲烷溶解,在氩气保护下,控温-5~0℃低温加入三溴化磷,将反应液室温反应14~18h后,将反应液倒入饱和碳酸氢钠溶液中,用二氯甲烷萃取,合并有机相,有机相用饱和氯化钠溶液洗,无水硫酸钠干燥后,蒸发溶剂后,过硅胶色谱柱纯化,得产物为4-(溴甲基)丙烯酸苯酯。
优选的,所述4-(羟甲基)丙烯酸苯酯与三溴化磷的摩尔比为1∶1.0~2.0。
优选的,所述步骤(3)的具体过程为:
将4-二乙氨基水杨醛与丙二酸二乙酯溶解于无水乙醇中,并加入哌啶作为催化剂,回流反应6~10h,反应完毕,蒸发溶剂后,加入冰醋酸和浓盐酸,回流反应6~10h,反应完毕,将反应液倒入冰水中,用40%氢氧化钠溶液调ph为5.0,有大量固体析出,并搅拌30~60min,减压过滤得到粗产物,用甲苯重结晶,得产物为7-二乙氨基香豆素。
优选的,所述4-二乙氨基水杨醛与丙二酸二乙酯的摩尔比为1∶2.0~2.5。
优选的,所述步骤(4)的具体过程为:
在氩气保护下,将三氯氧磷缓慢滴加至无水n,n-二甲基甲酰胺中,滴加完后室温反应30~60min得vilsmeier试剂,然后将7-二乙氨基香豆素用n,n-二甲基甲酰胺后缓慢滴加至上述反应液中,滴加完后,控温25~80℃反应12~18h,反应完毕,将反应液倒入冰水中,用20%氢氧化钠溶液调至有大量固体析出,减压过滤得到粗产物,用无水乙醇重结晶,得产物为7-二乙氨基香豆素-3-甲醛。
优选的,所述7-二乙氨基香豆素利用vilsmeier反应的温度为25~80℃。
优选的,所述步骤(5)的具体过程为:
将7-二乙氨基香豆素-3-甲醛和4-甲基吡啶溶于无水n,n-二甲基甲酰胺中,并加入对甲苯磺酸为脱水剂,回流反应4~8h,反应完毕,将反应液倒入冰水中,搅拌10~30min,有固体析出,减压过滤得到粗产物,过硅胶色谱柱纯化,得产物为7-二乙氨基-3-(4-吡啶基)乙烯基香豆素。
优选的,所述7-二乙氨基香豆素-3-甲醛与4-甲基吡啶的摩尔比为1∶1.0~2.0。
优选的,所述步骤(6)的具体过程为:
将4-(溴甲基)丙烯酸苯酯与7-二乙氨基-3-(4-吡啶基)乙烯基香豆素溶于乙腈中,加热至80~100℃反应6~12h,反应完毕,冷却至室温,加入石油醚有固体析出,减压过滤,得到式(1)所示的化合物。
优选的,所述4-(溴甲基)丙烯酸苯酯与7-二乙氨基-3-(4-吡啶基)乙烯基香豆素的摩尔比为1∶0.5~1.0。
作为一个总的发明构思,本发明还提供一种上述的检测半胱氨酸的新型荧光探针或上述的制备方法制得的检测半胱氨酸的新型荧光探针的应用,将所述半胱氨酸荧光探针与磷酸盐缓冲液(pbs)和二甲基亚砜(dmso)的缓冲液混合,再加入待测溶液中,得到混合溶液,利用两个不同发射波长处荧光强度变化来检测半胱氨酸的存在与否。
优选的,所述待测溶液中无半胱氨酸时,所述反应溶液的最大荧光发射波长位于644nm处,当加入半胱氨酸后,所述反应溶液的539nm处的荧光减弱,最大荧光发射波长蓝移至539nm处。
优选的,所述检测半胱氨酸的新型荧光探针检测半胱氨酸的检出浓度下限为:46.7nm。
与现有技术相比,本发明的优点在于:
1、本发明的一种检测半胱氨酸的新型荧光探针是以丙烯酰基为识别单元的荧光探针,实践表明,本发明的荧光探针分子在对半胱氨酸进行检测时表现出了较高的选择性和灵敏度。
3、本发明的一种检测半胱氨酸的新型的制备方法,关键中间体4-(溴甲基)丙烯酸苯酯为首次报道,且后处理过程简单,适于工业化生产。
附图说明
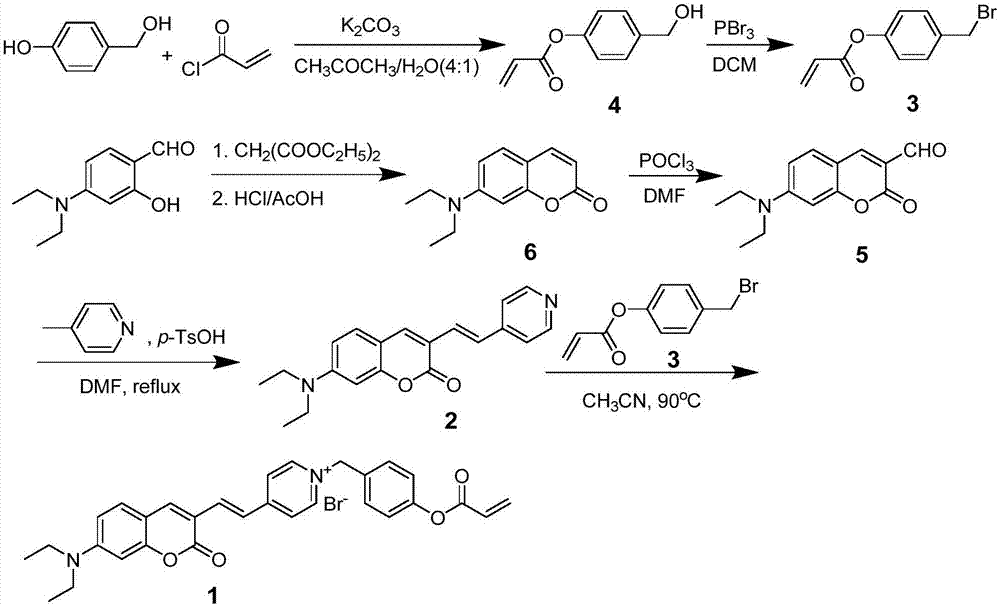
图1为实施例1的式(1)所示的化合物(即检测半胱氨酸的新型荧光探针)的合成路线图。
图2为实施例1制备的式(3)所示的化合物的1hnmr图谱。
图3为实施例1制备的式(3)所示的化合物的13cnmr图谱。
图4为实施例1制备的式(3)所示的化合物的esi-ms图谱。
图5为实施例1制备的式(1)所示的化合物的1hnmr图谱。
图6为实施例1制备的式(1)所示的化合物的13cnmr图谱。
图7为实施例1制备的式(1)所示的化合物的esi-ms图谱。
图8为不同ph值对实施例1制备的式(1)所示的化合物和式(2)所示的化合物的荧光强度比率(f539nm/f644nm)的影响((a)图)和不同ph值对实施例1制备的式(1)所示的化合物与cys反应前后荧光强度比率(f539nm/f644nm)的影响((b)图)。
图9为式(2)所示的化合物、式(1)所示的化合物与cys反应前后的吸收光谱((a)图)和荧光光谱图((b)图)。
图10为实施例1的式(1)所示的化合物与cys反应的荧光光谱图;其中,(a)图为(1)所示的化合物与cys反应后不同时间条件下荧光光谱的变化,(b)图为式(1)所示的化合物与cys反应荧光强度比率(f539nm/f644nm)随时间的变化。
图11为实施例1的式(1)所示的化合物(即检测半胱氨酸的新型荧光探针)对半胱氨酸检测的反应机理图。
图12为实施例1的式(1)所示的化合物分别与各种氨基酸和常见金属离子反应后荧光强度比率(f539nm/f644nm);
图13为实施例1的式(1)所示的化合物分别与各种氨基酸和常见金属离子反应后的荧光颜色。
图14为不同cys浓度对实施例1的式(1)所示的化合物与cys反应后荧光强度比率(f539nm/f644nm)的影响示意图;其中(a)图为不同cys浓度条件下,式(1)所示的化合物与cys反应后荧光强度比率(f539nm/f644nm)随时间的变化,(b)图为式(1)所示的化合物荧光强度比率(f539nm/f644nm)随cys浓度的变化。
具体实施方式
以下结合具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
实施例1:
一种本发明的检测半胱氨酸的新型荧光探针,名称为4-(7-二乙氨基香豆素-3-乙烯
基)-1-(4-丙烯酰氧基)苄基吡啶溴盐,分子式为c30h29brn2o4,结构式如式(1)所示:
上述本实施例的检测半胱氨酸的新型的制备方法,其合成路线如图1所示,包括以下步骤:
(1)合成结构式如式(4)所示的4-(羟甲基)丙烯酸苯酯:
反应式如式(3):
具体过程为:将碳酸钾(207mg,1.5mmol)溶于丙酮和水的混合溶液(25ml)中(体积比4:1),再加入丙烯酰氯(90.5mg,1mmol),冷却至0℃,控温0℃缓慢滴加对羟基苄醇(124mg,1mmol)溶液,滴加完后控温0℃反应4~6h,反应完后,用饱和氯化钠溶液淬灭,用二氯甲烷萃取,有机相用无水硫酸钠干燥,蒸发溶剂后,过硅胶色谱柱纯化,得产物为4-(羟甲基)丙烯酸苯酯,产率为40%。
(2)合成结构式如式(3)所示的4-(溴甲基)丙烯酸苯酯:
反应式如式(4):
具体过程为:将4-(羟甲基)丙烯酸苯酯(89.1mg,0.5mmol)用二氯甲烷(10ml)溶解,在氩气保护下,控温-5~0℃加入三溴化磷(0.05ml),将反应液室温反应14~18h后,将反应液倒入饱和碳酸氢钠溶液中,用二氯甲烷萃取,合并有机相,有机相用饱和氯化钠溶液洗,无水硫酸钠干燥后,蒸发溶剂后,过硅胶色谱柱纯化,得产物为4-(溴甲基)丙烯酸苯酯,产率为50%。
所得的4-(溴甲基)丙烯酸苯酯的1hnmr图谱如图2所示,1hnmr(400mhz,cdcl3,ppm)δ7.43(d,j=8.4hz,2h),7.13(d,j=8.8hz,2h),6.63-6.59(m,1h),6.35-6.28(m,1h),6.04-6.01(m,1h),4.49(s,2h)。
所得的4-(溴甲基)丙烯酸苯酯的13cnmr图谱如图3所示,13cnmr(100mhz,cdcl3,ppm)δ164.39,150.60,135.50,132.86,130.31,127.88,121.99,32.74。
所得的4-(溴甲基)丙烯酸苯酯的esi-ms图谱如图4所示,esi-msm/zforc10h9bro2+(m+):calcd:241.0,found:241.0。
(3)合成结构式如式(6)所示的7-二乙氨基香豆素:
反应式如式(5):
具体过程为:将4-二乙氨基水杨醛(3.86g,20mmol)与丙二酸二乙酯(6.4g,40mmol)溶解于无水乙醇(60ml)中,并加入哌啶(2ml)作为催化剂,回流反应6~10h,反应完毕,蒸发溶剂后,加入冰醋酸(40ml)和浓盐酸(40ml),回流反应6~10h,反应完毕,将反应液倒入冰水中,用40%氢氧化钠溶液调ph为5.0,有大量固体析出,并搅拌30~60min,减压过滤得到粗产物,用甲苯重结晶,得产物为7-二乙氨基香豆素,产率为72%。
(4)合成结构式如式(5)所示的7-二乙氨基香豆素-3-甲醛:
反应式如式(6):
具体过程为:在氩气保护下,将三氯氧磷(2.4ml)缓慢滴加至无水n,n-二甲基甲酰胺(2.4ml)中,滴加完后室温反应30~60min得vilsmeier试剂,然后将7-二乙氨基香豆素(1.8g,8.3mmol)用n,n-二甲基甲酰胺(15ml)后缓慢滴加至上述反应液中,滴加完后,控温25~80℃反应12~18h,反应完毕,将反应液倒入冰水中,用20%氢氧化钠溶液调至有大量固体析出,减压过滤得到粗产物,用无水乙醇重结晶,得产物为7-二乙氨基香豆素-3-甲醛,产率为70%。
(5)合成结构式如式(2)所示的7-二乙氨基-3-(4-吡啶基)乙烯基香豆素:
反应式如式(7):
具体过程为:将7-二乙氨基香豆素-3-甲醛(981.2mg,4mmol)和4-甲基吡啶(447mg,4.8mmol)溶于无水n,n-二甲基甲酰胺(20ml)中,并加入对甲苯磺酸(1.94g,11.2mmol)为脱水剂,回流反应4~8h,反应完毕,将反应液倒入冰水中,搅拌10~30min,有固体析出,减压过滤得到粗产物,过硅胶色谱柱纯化,得产物为7-二乙氨基-3-(4-吡啶基)乙烯基香豆素,产率为30%。
(6)合成结构式如式(1)所示的4-(7-二乙氨基香豆素-3-乙烯基)-1-(4-丙烯酰氧基)苄基吡啶溴盐:
反应式如式(8):
具体过程为:将4-(溴甲基)丙烯酸苯酯(72.3mg,0.3mmol)与7-二乙氨基-3-(4-吡啶基)乙烯基香豆素(73.7mg,0.23mmol)溶于乙腈(5ml)中,加热至80~100℃反应6~12h,反应完毕,冷却至室温,加入石油醚有固体析出,减压过滤,即式(1)所示的化合物,产率为72.7%。
式(1)所示的化合物的1hnmr图谱如图5所示,1hnmr(400mhz,d6-dmso,ppm)δ9.06(d,j=6.0hz,2h),8.25(s,1h),8.20(d,j=6.0hz,2h),7.88(d,j=16.0hz,1h),7.68-7.63(m,3h),7.53(d,j=8.8hz,1h),7.29(d,j=8.4hz,2h),6.78(d,j=8.4hz,1h),6.57-6.51(m,2h),6.43-6.37(m,1h),6.17(d,j=10.4hz,1h),5.78(s,2h),3.47-3.45(m,4h),1.15-1.11(m,6h)。
式(1)所示的化合物的13cnmr图谱如图6所示,13cnmr(100mhz,d6-dmso,ppm)δ163.94,159.51,156.31,153.76,151.98,150.74,145.55,143.93,137.38,133.85,132.31,130.75,130.07,127.43,123.64,122.49,122.45,113.59,110.02,108.38,96.19,61.19,44.34,12.34。
式(1)所示的化合物的esi-ms图谱如图7所示,esi-msm/zforc30h29n2o4+([m-br]+):calcd:481.3,found:481.1。
上述本实施例制得的检测半胱氨酸的新型荧光探针的应用,将10mmpbs/dmso体积比为1∶1,ph7.4的缓冲溶液2ml加入比色皿中,加入本实施例制得的检测半胱氨酸的新型荧光探针混合均匀后,再加入待测溶液进行测试,该待测溶液中无半胱氨酸时,所述反应溶液的最大荧光发射波长位于644nm处,当加入半胱氨酸后,所述反应溶液的644nm处的荧光减弱,最大荧光发射波长蓝移至539nm处。
上述本实施例制得的检测半胱氨酸的新型荧光探针的应用研究:
1、ph值对式(1)所示的化合物和式(2)所示的化合物的荧光强度比率(f539nm/f644nm)的影响
取实施例1合成的式(1)所示的化合物和式(2)所示的化合物溶于二甲基亚砜中,分别制成2mmol/l的储备液。当ph值不同时,在室温下以500nm为激发光测量式(1)所示的化合物和式(2)所示的化合物的荧光性质,结果如图8所示。实验结果表明,图(a)式(1)所示的化合物在生理ph4.0~10.0范围内荧光强度比率(f539nm/f644nm)几乎不变,而式(2)所示的化合物在生理ph5.0~10.0范围内荧光强度比率(f539nm/f644nm)几乎不变;图(b)式(1)所示的化合物与cys反应时荧光强度比率(f539nm/f644nm)在ph6.0~7.4范围变化明显,当ph7.4时变化最大,ph7.4~10.0范围内几乎保持不变。
2、式(2)所示的化合物和式(1)所示的化合物与cys反应的吸收光谱和荧光光谱研究
在最佳测试条件下,即10mmpbs,h2o/dmso为1∶1,v/v,ph7.4,25℃,研究式(2)所示的化合物和式(1)所示的化合物与半胱氨酸反应的吸收光谱和荧光光谱性质,结果如图9所示。图9中(a)图为式(2)所示的化合物和式(1)所示的化合物与半胱氨酸反应前后的吸收光谱图,(b)图为式(2)所示的化合物和式(1)所示的化合物与半胱氨酸反应前后的荧光光谱图。化合物1与cys反应后不同时间条件下荧光强度比率(f539nm/f644nm)的变化,如图10(a)所示;化合物1与cys反应荧光强度比率(f539nm/f644nm)随时间的变化,如图10(b)所示。实验结果证明,式(1)所示的化合物适用于cys的快速检测,原理如图11所示,cys通过亲核加成-分子内环化和随后的对羟基苄基1,6-消除反应,使丙烯酰基从式(1)所示的化合物上脱去,从而生成7-二乙氨基-3-(4-吡啶基)乙烯基香豆素。
3、式(1)所示的化合物对半胱氨酸的选择性研究
为测试式(1)所示的化合物对半胱氨酸的选择性,对式(1)所示的化合物与各种氨基酸和常见金属离子:gly,ser,leu,glu,pro,asn,phe,met,na+,k+,ca2+,mg2+,cys,gsh,hcy,反应后荧光强度比率(f539nm/f644nm)和荧光颜色变化进行了实验研究,结果如图12和图13所示。由图12和图13可知,式(1)所示的化合物对半胱氨酸的检测具有高度的选择性。
4、式(1)所示的化合物检测半胱氨酸的灵敏度研究
不同半光氨酸浓度0-80μm条件下,式(1)所示的化合物与半胱氨酸反应后荧光强度比率(f539nm/f644nm)随时间的变如图14(a)所示,式(1)所示的化合物荧光强度比率(f539nm/f644nm)随半胱氨酸浓度的变化如图14(b)可知,高浓度半胱氨酸提供更快和更戏剧性的荧光强度比率(f539nm/f644nm)增大。由图14(b)可以推算出式(1)所示的化合物检测半胱氨酸的检测限为46.7nm。
以上所述,仅是本申请的较佳实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
- 还没有人留言评论。精彩留言会获得点赞!